


Genes 2024, 15(5), 563; https://0-doi-org.brum.beds.ac.uk/10.3390/genes15050563 (registering DOI) - 27 Apr 2024
Abstract
TR2 and TR4 (NR2C1 and NR2C2, respectively) are evolutionarily conserved nuclear orphan receptors capable of binding direct repeat sequences in a stage-specific manner. Like other nuclear receptors, TR2 and TR4 possess important roles in transcriptional activation or repression with developmental stage and tissue
[...] Read more.
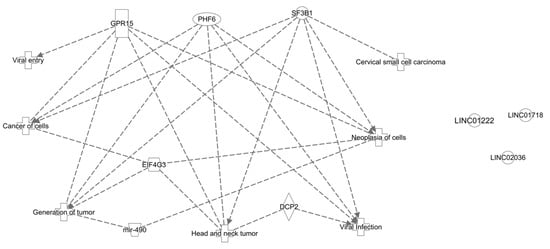
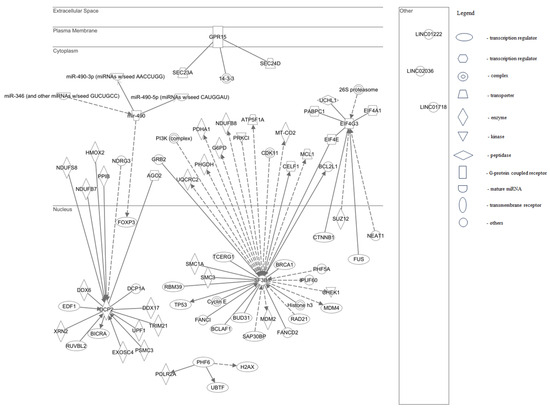
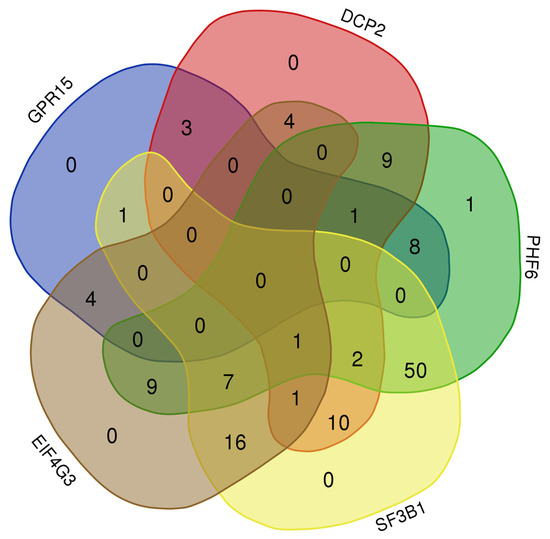
TR2 and TR4 (NR2C1 and NR2C2, respectively) are evolutionarily conserved nuclear orphan receptors capable of binding direct repeat sequences in a stage-specific manner. Like other nuclear receptors, TR2 and TR4 possess important roles in transcriptional activation or repression with developmental stage and tissue specificity. TR2 and TR4 bind DNA and possess the ability to complex with available cofactors mediating developmental stage-specific actions in primitive and definitive erythrocytes. In erythropoiesis, TR2 and TR4 are required for erythroid development, maturation, and key erythroid transcription factor regulation. TR2 and TR4 recruit and interact with transcriptional corepressors or coactivators to elicit developmental stage-specific gene regulation during hematopoiesis.
Full article
(This article belongs to the Section Molecular Genetics and Genomics)









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}