
Gender Impacted Gut Microbiota and Growth Performance in the Blotched Snakehead (Channa maculata)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Fish and Sample Collection
2.2. High-Throughput Sequencing
2.3. Bioinformatic and Statistical Analysis
3. Results
3.1. Growth Performance Analysis
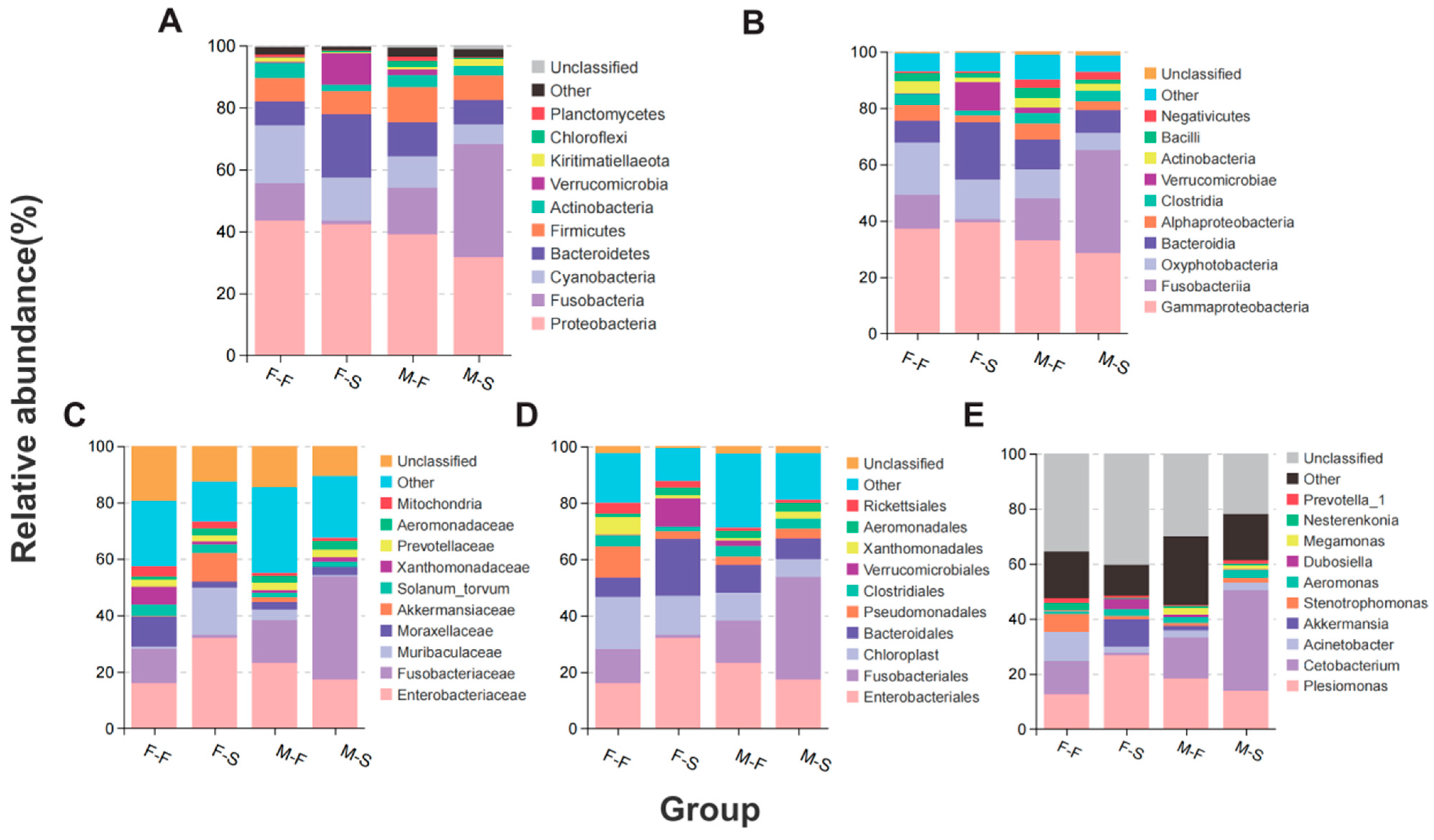
3.2. Microbial Taxonomic Composition
3.3. Alpha and Beta Diversity of Gut Microbiota
3.4. Linking Growth Performance and Gender to Biomarkers
3.5. Structure of Potential Metabolic Function and Microbial Network
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gong, L.-C.; Wang, H.; Deng, L. Molecular characterization, phylogeny and expression of a hepcidin gene in the blotched snakehead Channa maculata. Dev. Comp. Immunol. 2014, 44, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Huang, Y.; Xie, J.; Wang, G.; Yu, D.; Sun, X.; Zhang, K.; Li, Z.; Ermeng, Y.; Tian, J.; et al. Identification and expression analysis of miRNA in hybrid snakehead by deep sequencing approach and their targets prediction. Genomics 2019, 111, 1315–1324. [Google Scholar] [CrossRef]
- Zhou, A.; Xie, S.; Wang, Z.; Junaid, M.; Fan, L.; Wang, C.; Ye, Q.; Chen, Y.; Pei, D.S.; Zou, J. Molecular cloning, characterization and expression analysis of heat shock protein 90 in albino northern snakehead Channa argus. Gene 2017, 626, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.M.; Xu, M.X.; Lin, S.M.; Liang, Z.; Cai, J.H. Association of cyclin D1 and survivin expression with sensitivity to radiotherapy in patients with nasopharyngeal carcinoma. Genet. Mol. Res. 2014, 13, 3502–3509. [Google Scholar] [CrossRef]
- Yearbook, C.F.S. China Fishery Statistical Yearbook; China Agriculture Press: Beijing, China, 2020; Volume 2020, pp. 24–34. [Google Scholar]
- Zhao, J.; Ou, M.; Wang, Y.; Liu, H.; Luo, Q.; Zhu, X.; Chen, B.; Chen, K. Breeding of YY super-male of blotched snakehead (Channa maculata) and production of all-male hybrid (Channa argus ♀ × C. maculata ♂). Aquaculture 2021, 538, 736450. [Google Scholar] [CrossRef]
- Prchalová, M.; Žák, J.; Říha, M.; Šmejkal, M.; Blabolil, P.; Vašek, M.; Matěna, J.; Peterka, J.; Seďa, J.; Kubečka, J. Sexual size dimorphism of two common European percid fish: Linkage with spatial distribution and diet. Hydrobiologia 2022, 849, 2009–2027. [Google Scholar] [CrossRef]
- Chen, S.-L.; Ji, X.-S.; Shao, C.-W.; Li, W.-L.; Yang, J.-F.; Liang, Z.; Liao, X.L.; Xu, G.B.; Xu, Y.; Song, W.-T. Induction of Mitogynogenetic Diploids and Identification of WW Super-female Using Sex-Specific SSR Markers in Half-Smooth Tongue Sole (Cynoglossus semilaevis). Mar. Biotechnol. 2011, 14, 120–128. [Google Scholar] [CrossRef]
- Dan, C.; Mei, J.; Wang, D.; Gui, J.-F. Genetic Differentiation and Efficient Sex-specific Marker Development of a Pair of Y- and X-linked Markers in Yellow Catfish. Int. J. Biol. Sci. 2013, 9, 1043–1049. [Google Scholar] [CrossRef]
- Ou, M.; Yang, C.; Luo, Q.; Huang, R.; Zhang, A.D.; Liao, L.J.; Li, Y.M.; He, L.B.; Zhu, Z.Y.; Chen, K.C.; et al. An NGS-based approach for the identification of sex-specific markers in snakehead (Channa argus). Oncotarget 2017, 8, 98733. [Google Scholar] [CrossRef]
- Dawood, M.A.O. Nutritional immunity of fish intestines: Important insights for sustainable aquaculture. Rev. Aquac. 2020, 13, 642–663. [Google Scholar] [CrossRef]
- Li, H.; Lu, L.; Chen, R.; Li, S.; Xu, D. Exploring Sexual Dimorphism in the Intestinal Microbiota of the Yellow Drum (Nibea albiflora, Sciaenidae). Front. Microbiol. 2022, 12, 808285. [Google Scholar] [CrossRef]
- Gruneck, L.; Jinatham, V.; Therdtatha, P.; Popluechai, S. Siamese Fighting Fish (Betta splendens Regan) Gut Microbiota Associated with Age and Gender. Fishes 2022, 7, 347. [Google Scholar] [CrossRef]
- Navarro-Barrón, E.; Hernández, C.; Llera-Herrera, R.; García-Gasca, A.; Gómez-Gil, B. Overfeeding a High-Fat Diet Promotes Sex-Specific Alterations on the Gut Microbiota of the Zebrafish (Danio rerio). Zebrafish 2019, 16, 268–279. [Google Scholar] [CrossRef]
- Chen, Z.-W.; Jin, X.-K.; Gao, F.-X.; Gui, J.-F.; Zhao, Z.; Shi, Y. Comparative analyses reveal sex-biased gut microbiota in cultured subadult pufferfish Takifugu obscurus. Aquaculture 2022, 558, 738366. [Google Scholar] [CrossRef]
- Li, X.; Yan, Q.; Ringø, E.; Wu, X.; He, Y.; Yang, D. The influence of weight and gender on intestinal bacterial community of wild largemouth bronze gudgeon (Coreius guichenoti, 1874). BMC Microbiol. 2016, 16, 191. [Google Scholar] [CrossRef]
- Li, M.; Zhu, X.; Tian, J.; Liu, M.; Wang, G. Bioaccumulation, oxidative stress, immune responses and immune-related genes expression in northern snakehead fish, Channa argus, exposure to waterborne selenium. Mol. Biol. Rep. 2018, 46, 947–955. [Google Scholar] [CrossRef]
- Han, C.; Zhou, X.; Lu, H.; Zhu, Q.; Han, L.; Li, S.; Lin, H.; Zhang, Y. A simple PCR-based genetic sex identification method in the blotched snakehead (Channa maculata) developed by high-throughput sequencing. Aquaculture 2021, 538, 736579. [Google Scholar] [CrossRef]
- Feng, Q.-M.; Ru, X.-S.; Zhang, L.-B.; Zhang, S.-Y.; Yang, H.-S. Differences in feeding behavior and intestinal microbiota may relate to different growth rates of sea cucumbers (Apostichopus japonicus). Aquaculture 2022, 559, 738368. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Fu, B.; Fang, C.; Xia, J.; Pan, S.; Zhou, L.; Peng, Y.; Yan, Y.; Yang, Y.; He, Y.; Chen, S.; et al. Urbanization alters soil bacterial communities in southern China coastal cities. Ecotoxicol. Environ. Saf. 2023, 250, 114492. [Google Scholar] [CrossRef]
- Fang, C.; He, Y.; Yang, Y.; Fu, B.; Pan, S.; Jiao, F.; Wang, J.; Yang, H. Laboratory tidal microcosm deciphers responses of sediment archaeal and bacterial communities to microplastic exposure. J. Hazard. Mater. 2023, 458, 131813. [Google Scholar] [CrossRef]
- Nayak, S.K. Role of gastrointestinal microbiota in fish. Aquac. Res. 2010, 41, 1553–1573. [Google Scholar] [CrossRef]
- Chang, S.; Wang, J.; Dong, C.; Jiang, Y. Intestinal microbiota signatures of common carp (Cyprinus carpio) after the infection of Aeromonas hydrophila. Aquac. Rep. 2023, 30, 101585. [Google Scholar] [CrossRef]
- Ye, L.; Liu, G.; Yao, T.; Lu, J. Monitoring of antimicrobial resistance genes in the spotted sea bass (Lateolabrax maculatus): Association with the microbiome and its environment in aquaculture ponds. Environ. Pollut. 2021, 276, 116714. [Google Scholar] [CrossRef]
- Yuan, L.; Wang, L.; Li, Z.-H.; Zhang, M.-Q.; Shao, W.; Sheng, G.-P. Antibiotic resistance and microbiota in the gut of Chinese four major freshwater carp from retail markets. Environ. Pollut. 2019, 255, 113327. [Google Scholar] [CrossRef] [PubMed]
- Tarnecki, A.M.; Burgos, F.A.; Ray, C.L.; Arias, C.R. Fish intestinal microbiome: Diversity and symbiosis unravelled by metagenomics. J. Appl. Microbiol. 2017, 123, 2–17. [Google Scholar] [CrossRef]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The Effects of Vegetarian and Vegan Diets on Gut Microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef]
- Pan, X.; Raaijmakers, J.M.; Carrión, V.J. Importance of Bacteroidetes in host–microbe interactions and ecosystem functioning. Trends Microbiol. 2023, 31, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, C.; Sakata, T.; Sugita, H. Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Lett. Appl. Microbiol. 2007, 46, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Sugita, H.; Miyajima, C.; Deguchi, Y. The vitamin B12-producing ability of the intestinal microflora of freshwater fish. Aquaculture 1991, 92, 267–276. [Google Scholar] [CrossRef]
- Liao, X.; Zhao, P.; Hou, L.; Adyari, B.; Xu, E.G.; Huang, Q.; Hu, A. Network analysis reveals significant joint effects of microplastics and tetracycline on the gut than the gill microbiome of marine medaka. J. Hazard. Mater. 2023, 442, 129996. [Google Scholar] [CrossRef]
- Eduardo, C.I.-A.; Romo-Vaquero, M.; Victoria, M.S.; Carlos, J.E. Unveiling metabotype clustering in resveratrol, daidzein, and ellagic acid metabolism: Prevalence, associated gut microbiomes, and their distinctive microbial networks. Food Res. Int. 2023, 173, 113470. [Google Scholar] [CrossRef]
- Wu, Z.; Kang, J.; Zhang, C.; Zhang, W.; Ge, J. Assessing the promoting effect of compound microbial agents on flax dew retting: Based on the relationship between metabolites and core genera. Bioresour. Technol. 2023, 385, 129451. [Google Scholar] [CrossRef]
- Siddle, K.J.; Quintana-Murci, L. The Red Queen’s long race: Human adaptation to pathogen pressure. Curr. Opin. Genet. Dev. 2014, 29, 31–38. [Google Scholar] [CrossRef]
- Ye, G.; Dong, X.; Yang, Q.; Chi, S.; Liu, H.; Zhang, H.; Tan, B.; Zhang, S. Low-gossypol cottonseed protein concentrate used as a replacement of fish meal for juvenile hybrid grouper (Epinephelus fuscoguttatus ♀ × Epinephelus lanceolatus ♂): Effects on growth performance, immune responses and intestinal microbiota. Aquaculture 2020, 524, 735309. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Dai, J.; Yang, P.; Xu, W.; Ai, Q.; Zhang, W.; Zhang, Y.; Zhang, Y.; Mai, K. Sodium butyrate supplementation in high-soybean meal diets for turbot (Scophthalmus maximus L.): Effects on inflammatory status, mucosal barriers and microbiota in the intestine. Fish Shellfish Immunol. 2019, 88, 65–75. [Google Scholar] [CrossRef]
- Tian, B.; Hua, Y. Carotenoid biosynthesis in extremophilic Deinococcus–Thermus bacteria. Trends Microbiol. 2010, 18, 512–520. [Google Scholar] [CrossRef]
- Luo, K.; Xiao, J.; Liu, S.; Wang, J.; He, W.; Hu, J.; Qin, Q.; Zhang, C.; Tao, M.; Liu, Y. Massive production of all-female diploids and triploids in the crucian carp. Int. J. Biol. Sci. 2011, 7, 487. [Google Scholar] [CrossRef]
- Wu, L.; Xu, Y.; Lv, X.; Chang, X.; Ma, X.; Tian, X.; Shi, X.; Li, X.; Kong, X. Impacts of an azo food dye tartrazine uptake on intestinal barrier, oxidative stress, inflammatory response and intestinal microbiome in crucian carp (Carassius auratus). Ecotoxicol. Environ. Saf. 2021, 223, 112551. [Google Scholar] [CrossRef]
- Ramirez-Farias, C.; Slezak, K.; Fuller, Z.; Duncan, A.; Holtrop, G.; Louis, P. Effect of inulin on the human gut microbiota: Stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br. J. Nutr. 2008, 101, 541–550. [Google Scholar] [CrossRef]
- Kong, C.; Gao, R.; Yan, X.; Huang, L.; Qin, H. Probiotics improve gut microbiota dysbiosis in obese mice fed a high-fat or high-sucrose diet. Nutrition 2019, 60, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, A.F.; Sheng, N.; Nicol, K.; Strömberg, N.; Hollox, E.J. Balancing selection at the human salivary agglutinin gene (DMBT1) driven by host-microbe interactions. iScience 2022, 25, 104189. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Peng, L.; Feng, P.; Han, R.; Khan, A.; Kulshreshtha, S.; Ling, Z.; Liu, P.; Li, X. Gut microbes consume host energy and reciprocally provide beneficial factors to sustain a symbiotic relationship with the host. Sci. Total Environ. 2023, 904, 166773. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Yin, S.; Qin, G.; Yao, J.; Liu, S.; Han, J.; Zhou, Y.; Duan, S. Gastrointestinal digestion and absorption of soybean β-conglycinin in an early weaned piglet model: An initial step to the induction of soybean allergy. Food Chem. 2023, 427, 136640. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wang, Y.; Wang, C.; Zhang, L.; Tu, K.; Zheng, Z. Insights into the intestinal microbiota of several aquatic organisms and association with the surrounding environment. Aquaculture 2019, 507, 196–202. [Google Scholar] [CrossRef]
- Zanello, P. The competition between chemistry and biology in assembling iron–sulfur derivatives. Molecular structures and electrochemistry. Part V. {[Fe4S4](SCysγ)4} proteins. Coord. Chem. Rev. 2017, 335, 172–227. [Google Scholar] [CrossRef]
- Teklebrhan, T.; Tan, Z.; Jonker, A. Diet containing sulfur shifted hydrogen metabolism from methanogenesis to alternative sink and influenced fermentation and gut microbial ecosystem of goats. Anim. Feed Sci. Technol. 2022, 294, 115480. [Google Scholar] [CrossRef]
- Uchiyama, J.; Akiyama, M.; Hase, K.; Kumagai, Y.; Kim, Y.-G. Gut microbiota reinforce host antioxidant capacity via the generation of reactive sulfur species. Cell Rep. 2022, 38, 110479. [Google Scholar] [CrossRef]
- MacPherson, J.; Weinrauch, A.M.; Anderson, W.G.; Bucking, C. The gut microbiome may influence post-prandial nitrogen handling in an elasmobranch, the Pacific spiny dogfish (Squalus suckleyi). Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2022, 272, 111269. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, J.T.; Monticelli, G.; Schlenk, D.; Bisesi, J.H., Jr.; Pampanin, D.M. Connecting gut microbiome changes with fish health conditions in juvenile Atlantic cod (Gadus morhua) exposed to dispersed crude oil. Environ. Res. 2023, 234, 116516. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, B.; Cai, L.; Kang, J.; Dong, Z.; Zhang, B.; Wang, B.; Gong, Y.; Xu, Z.; Zhang, D.; et al. Comparative analysis of oxidized fish oil and coenzyme Q10 on the intestinal microecology of largemouth bass Micropterus salmoides at different growth stages. Aquac. Rep. 2023, 30, 101608. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group (Mean ± SE) | ||||
|---|---|---|---|---|
| F-F (n = 9) | F-S (n = 9) | M-F (n = 9) | M-S (n = 9) | |
| Total length (cm) | 32.33 ± 0.76 a | 24.30 ± 0.62 b | 34.93 ± 1.01 a | 27.40 ± 4.26 b |
| Height (cm) | 6.43 ± 0.06 a | 5.13 ± 0.42 b | 6.70 ± 0.44 a | 5.37 ± 0.65 b |
| Standard length (cm) | 27.90 ± 1.15 a | 20.90 ± 0.56 b | 30.70 ± 0.75 a | 23.53 ± 3.29 b |
| Weight (g) | 424.07 ± 38.13 a | 184.97 ± 25.02 b | 540.43 ± 50.34 a | 262.50 ± 83.75 b |
| Group (Mean ± SE) | F | p | ||||
|---|---|---|---|---|---|---|
| F-F (n = 3) | F-S (n = 3) | M-F (n = 3) | M-S (n = 3) | |||
| Nodes | 56.67 ± 0.58 | 56.00 ± 1.73 | 56.00 ± 1.00 | 55.33 ± 2.89 | 0.281 | 0.838 |
| Edges | 220.67 ± 5.77 | 211.00 ± 22.52 | 219.00 ± 7.81 | 222.67 ± 2.31 | 0.517 | 0.682 |
| Edge density | 0.14 ± 0.00 | 0.14 ± 0.01 | 0.14 ± 0.01 | 0.15 ± 0.01 | 1.036 | 0.427 |
| Degree centralization | 0.14 ± 0.01 | 0.14 ± 0.01 | 0.14 ± 0.01 | 0.15 ± 0.00 | 0.488 | 0.700 |
| Betweenness centralization | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 1.484 | 0.291 |
| Average clustering coefficient | 0.93 ± 0.01 | 0.92 ± 0.00 | 0.93 ± 0.00 | 0.93 ± 0.00 | 1.169 | 0.380 |
| Complexity | 3.89 ± 0.06 | 3.76 ± 0.29 | 3.91 ± 0.15 | 4.03 ± 0.17 | 1.018 | 0.434 |
| Modularity | 0.62 ± 0.01 | 0.62 ± 0.02 | 0.62 ± 0.01 | 0.61 ± 0.01 | 0.690 | 0.583 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, C.; Zeng, F.; Chen, S.; Li, S.; Yang, Y.; Lin, W.; Liu, Y.; Peng, C.; Yang, H. Gender Impacted Gut Microbiota and Growth Performance in the Blotched Snakehead (Channa maculata). Microorganisms 2024, 12, 871. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms12050871
Fang C, Zeng F, Chen S, Li S, Yang Y, Lin W, Liu Y, Peng C, Yang H. Gender Impacted Gut Microbiota and Growth Performance in the Blotched Snakehead (Channa maculata). Microorganisms. 2024; 12(5):871. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms12050871
Chicago/Turabian StyleFang, Chang, Fang Zeng, Shijun Chen, Shuisheng Li, Yuting Yang, Wanjing Lin, Yun Liu, Cheng Peng, and Huirong Yang. 2024. "Gender Impacted Gut Microbiota and Growth Performance in the Blotched Snakehead (Channa maculata)" Microorganisms 12, no. 5: 871. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms12050871