
The Contributions of Sub-Communities to the Assembly Process and Ecological Mechanisms of Bacterial Communities along the Cotton Soil–Root Continuum Niche Gradient
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Experimental Design and Sample Collection
2.2. Extraction of Genome DNA and Amplicon Generation
2.3. PCR Products Quantification and Qualification
2.4. Library Preparation and Sequencing
2.5. Definition of Abundant Taxa, Moderate Taxa, and Rare Taxa
2.6. Calculations and Statistical Analyses
2.7. Co-Occurrence Network Analysis
3. Results
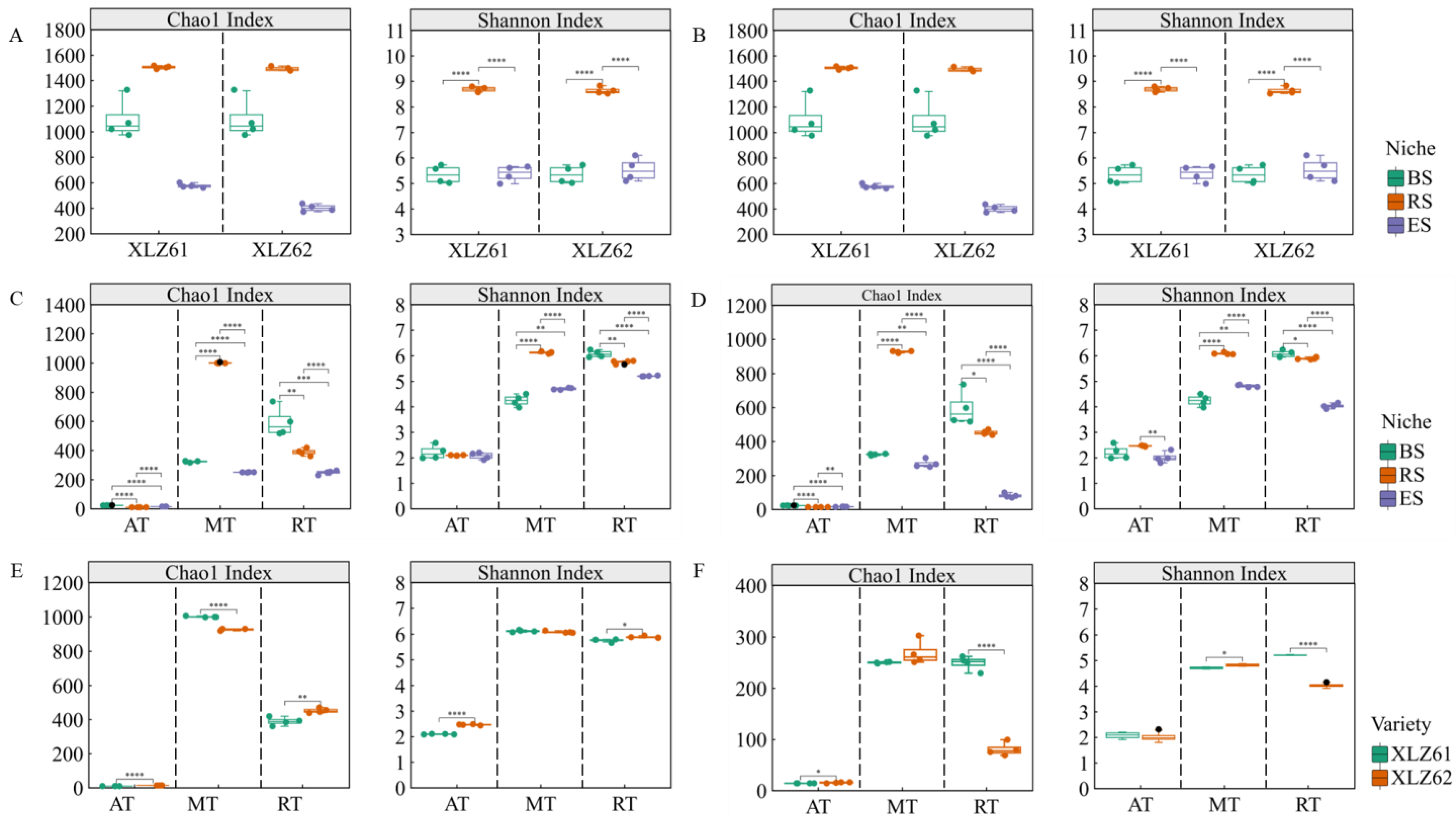
3.1. Differential Analysis of Bacterial Community Composition and Structure along the Cotton Soil–Root Continuum Compartment Niches
3.2. Differences in Bacterial Community Diversity along the Niche Gradient in the Cotton Soil–Root Continuum
3.3. Assembly Mechanisms of Microbial Communities along the Niche Gradient in the Soil–Root Continuum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Der Heijden, M.G.A.; Bardgett, R.D.; Van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.D.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Liu, W.; Qiu, X.; Zhang, J.; Zhang, J.; Bai, Y. The root microbiome: Community assembly and its contributions to plant fitness. J. Integr. Plant Biol. 2022, 64, 230–243. [Google Scholar] [CrossRef]
- Zhang, R.; Vivanco, J.M.; Shen, Q. The unseen rhizosphere root-soil-microbe interactions for crop production. Curr. Opin. Microbiol. 2017, 37, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Badri, D.V.; Vivanco, J.M. Regulation and function of root exudates. Plant Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Jiang, T.; Liu, Y.X.; Bai, Y.C.; Reed, J.; Qu, B.; Goossens, A.; Nützmann, H.W.; Bai, Y.; Osbourn, A. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 2019, 364, eaau6389. [Google Scholar] [CrossRef]
- Wang, N.R.; Haney, C.H. Harnessing the genetic potential of the plant microbiome. Biochemist 2020, 42, 20–25. [Google Scholar] [CrossRef]
- Huang, X.F.; Chaparro, J.M.; Reardon, K.F.; Zhang, R.; Shen, Q.; Vivanco, J.M. Rhizosphere interactions: Root exudates, microbes, and microbial communities. Botany 2014, 92, 267–275. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Ullah, A.; Akbar, A.; Luo, Q.; Khan, A.H.; Manghwar, H.; Shaban, M.; Yang, X. Microbiome diversity in cotton rhizosphere under normal and drought conditions. Microb. Ecol. 2019, 77, 429–439. [Google Scholar] [CrossRef]
- Chen, Y.; Ding, Q.; Chao, Y.; Wei, X.; Wang, S.; Qiu, R. Structural development and assembly patterns of the root-associated microbiomes during phytoremediation. Sci. Total Environ. 2018, 644, 1591–1601. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed]
- Beckers, B.; Op De Beeck, M.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Ver Loren van Themaat, E.V.L.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef]
- Hartman, K.; van der Heijden, M.G.A.; Wittwer, R.A.; Banerjee, S.; Walser, J.C.; Schlaeppi, K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 2018, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Deng, Y.; Zhang, P.; Xue, K.; Liang, Y.; Van Nostrand, J.D.; Yang, Y.; He, Z.; Wu, L.; Stahl, D.A.; et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc. Natl. Acad. Sci. USA 2014, 111, E836–E845. [Google Scholar] [CrossRef]
- Vorholt, J.A.; Vogel, C.; Carlström, C.I.; Müller, D.B. Establishing causality: Opportunities of synthetic communities for plant microbiome research. Cell Host Microbe 2017, 22, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; He, J.Z.; Singh, B.K.; Zhu, Y.G.; Wang, J.T.; Li, P.P.; Zhang, Q.B.; Han, L.L.; Shen, J.P.; Ge, A.H.; et al. Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ. Microbiol. 2021, 23, 1907–1924. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wei, G. Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 2017, 26, 5305–5317. [Google Scholar] [CrossRef]
- Ziegler, M.; Eguíluz, V.M.; Duarte, C.M.; Voolstra, C.R. Rare symbionts may contribute to the resilience of coral–algal assemblages. ISME J. 2018, 12, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Montoya, L.; Xu, L.; Madera, M.; Hollingsworth, J.; Purdom, E.; Singan, V.; Vogel, J.; Hutmacher, R.B.; Dahlberg, J.A.; et al. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat. Commun. 2020, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Lu, Y. Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob. Chang. Biol. 2020, 26, 4506–4520. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Liu, Z.; Lin, Y.; Yang, J.; Chen, W.; Wei, G. Bacterial communities in oil contaminated soils: Biogeography and co-occurrence patterns. Soil. Biol. Biochem. 2016, 98, 64–73. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhang, P.; Qin, Y.; Tu, Q.; Yang, Y.; He, Z.; Schadt, C.W.; Zhou, J. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environ. Microbiol. 2016, 18, 205–218. [Google Scholar] [CrossRef]
- Xiong, C.; Lu, Y. Microbiomes in agroecosystem: Diversity, function and assembly mechanisms. Environ. Microbiol. Rep. 2022, 14, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Mao, S.-C.; Tian, L.-W.; Li, L.; Dong, H.-Z. Advances and prospects of high-yielding and simplified cotton cultivation technology in Xinjiang cotton-growing area. Sci. Agric. Sin. 2017, 50, 38–50. [Google Scholar]
- Lou, S.-W.; Dong, H.-Z.; Tian, X.-L.; Tian, L.-W. The “short, dense and early” cultivation of cotton in Xinjiang: History, current situation and prospect. Sci. Agric. Sin. 2021, 54, 720–732. [Google Scholar]
- Qiao, Q.; Wang, F.; Zhang, J.; Chen, Y.; Zhang, C.; Liu, G.; Zhang, H.; Ma, C.; Zhang, J. The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage. Sci. Rep. 2017, 7, 3940. [Google Scholar] [CrossRef]
- Wei, F.; Zhao, L.; Xu, X.; Feng, H.; Shi, Y.; Deakin, G.; Feng, Z.; Zhu, H. Cultivar-dependent variation of the cotton rhizosphere and endosphere microbiome under field conditions. Front. Plant Sci. 2019, 10, 1659. [Google Scholar] [CrossRef]
- Wei, Z.; Gu, Y.; Friman, V.P.; Kowalchuk, G.A.; Xu, Y.; Shen, Q.; Jousset, A. Initial soil microbiome composition and functioning predetermine future plant health. Sci. Adv. 2019, 5, eaaw0759. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Yu, D. Rhizosphere fungal community structure succession of Xinjiang continuously cropped cotton. Fungal Biol. 2019, 123, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-J.; Liu, S.-D.; Liu, R.-H.; Zhang, S.-P.; Yang, J.; Pang, C.-Y. Effects of crop rotation on bacterial communities in cotton rhizosphere soil. Biotechnol. Bull. 2020, 36, 117–124. [Google Scholar]
- Zhong, Y.; Sorensen, P.O.; Zhu, G.; Jia, X.; Liu, J.; Shangguan, Z.; Wang, R.; Yan, W. Differential microbial assembly processes and co-occurrence networks in the soil-root continuum along an environmental gradient. iMeta 2022, 1, e18. [Google Scholar] [CrossRef]
- Liu, L.M.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The Vegan Package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Levins, R. Evolution in Changing Environments; Princeton University Press: Princeton, NJ, USA, 1968. [Google Scholar]
- Burns, A.R.; Stephens, W.Z.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J.M. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016, 10, 655–664. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: http://www.R-project.org (accessed on 15 February 2024).
- Pandit, S.N.; Kolasa, J.; Cottenie, K. Contrasts between habitat generalists and specialists: An empirical extension to the basic metacommunity framework. Ecology 2009, 90, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.M.; Wang, M.M.; Zhou, X.; Zhang, A.M.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 2018, 175, 973–983.e14. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Clément, C.; Sessitsch, A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil. Biol. Biochem. 2010, 42, 669–678. [Google Scholar] [CrossRef]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 2014, 5, e01371-14. [Google Scholar] [CrossRef]
- Siqueira, T.; Bini, L.M.; Roque, F.O.; Marques Couceiro, S.R.; Trivinho-Strixino, S.; Cottenie, K. Common and rare species respond to similar niche processes in macroinvertebrate metacommunities. Ecography 2012, 35, 183–192. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | BS | RS | ES | ||
|---|---|---|---|---|---|
| XLZ61 | XLZ62 | XLZ61 | XLZ62 | ||
| Total OTUs | 1428 (1) | 1645 (1) | 1660 (1) | 787 (1) | 611 (1) |
| AT OTUs | 23 (66.31%) | 9 (17.42%) | 13 (21.08%) | 14 (64.43%) | 16 (59.74%) |
| MT OTUs | 347 (30.26%) | 1010 (79.25%) | 941 (75.23%) | 252 (34.1%) | 347 (39.41%) |
| RT OTUs | 1058 (3.43%) | 626 (3.33%) | 706 (3.69%) | 521 (1.47%) | 248 (0.85%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Liu, R.; Zhang, S.; Shen, Q.; Chen, J.; Ma, H.; Ge, C.; Hao, L.; Zhang, J.; Shi, S.; et al. The Contributions of Sub-Communities to the Assembly Process and Ecological Mechanisms of Bacterial Communities along the Cotton Soil–Root Continuum Niche Gradient. Microorganisms 2024, 12, 869. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms12050869
Liu S, Liu R, Zhang S, Shen Q, Chen J, Ma H, Ge C, Hao L, Zhang J, Shi S, et al. The Contributions of Sub-Communities to the Assembly Process and Ecological Mechanisms of Bacterial Communities along the Cotton Soil–Root Continuum Niche Gradient. Microorganisms. 2024; 12(5):869. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms12050869
Chicago/Turabian StyleLiu, Shaodong, Ruihua Liu, Siping Zhang, Qian Shen, Jing Chen, Huijuan Ma, Changwei Ge, Lidong Hao, Jinshan Zhang, Shubing Shi, and et al. 2024. "The Contributions of Sub-Communities to the Assembly Process and Ecological Mechanisms of Bacterial Communities along the Cotton Soil–Root Continuum Niche Gradient" Microorganisms 12, no. 5: 869. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms12050869