
Gastric Inhibitory Polypeptide Receptor (GIPR) Overexpression Reduces the Tumorigenic Potential of Retinoblastoma Cells

 , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Retina and Retinoblastoma Samples
2.2. Human Cell Lines and Culture
2.3. Expression Vectors
2.4. Luciferase Binding Studies
2.5. Transient GIPR and miR-542-5p Overexpression
2.6. Lentivirus Production and Transduction
2.7. RNA Extraction and Quantitative Real-Time PCR
2.8. Western Blot Analyses
2.9. Cell Viability Assays
2.10. Growth Kinetics
2.11. BrdU and Caspase-3 Assays
2.12. GIPR Inhibitor Studies
2.13. In Ovo Chorioallantoic Membrane (CAM) Assays
2.14. Cancer-Related Protein Expression Profiling
2.15. Statistical Analysis
3. Results
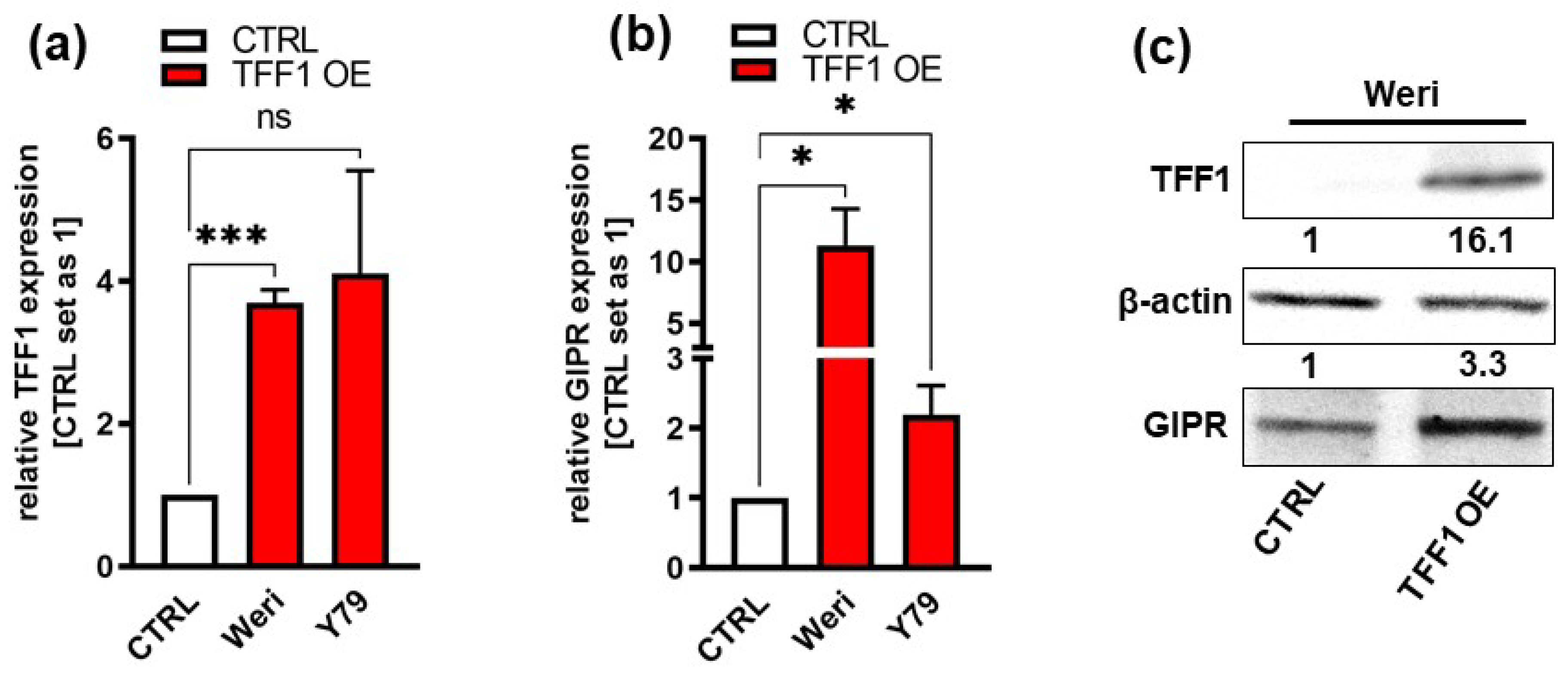
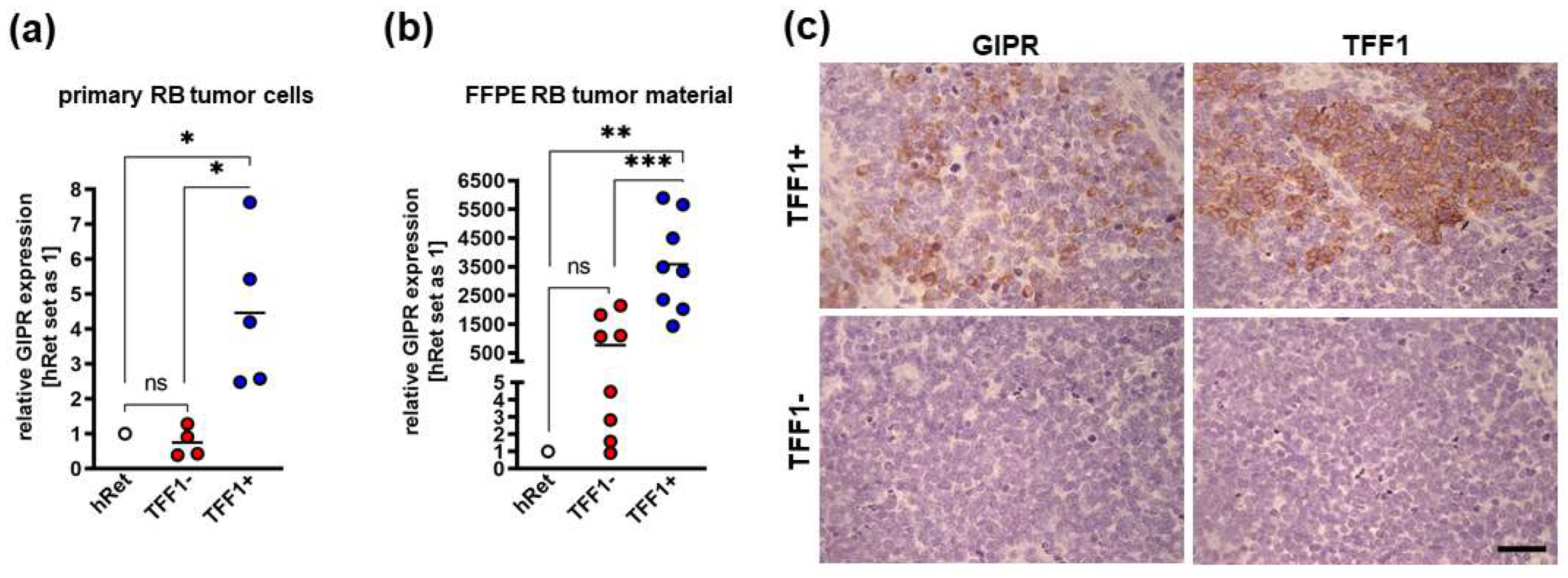
3.1. GIPR and TFF1 Are Co-Expressed in Retinoblastoma Tumors
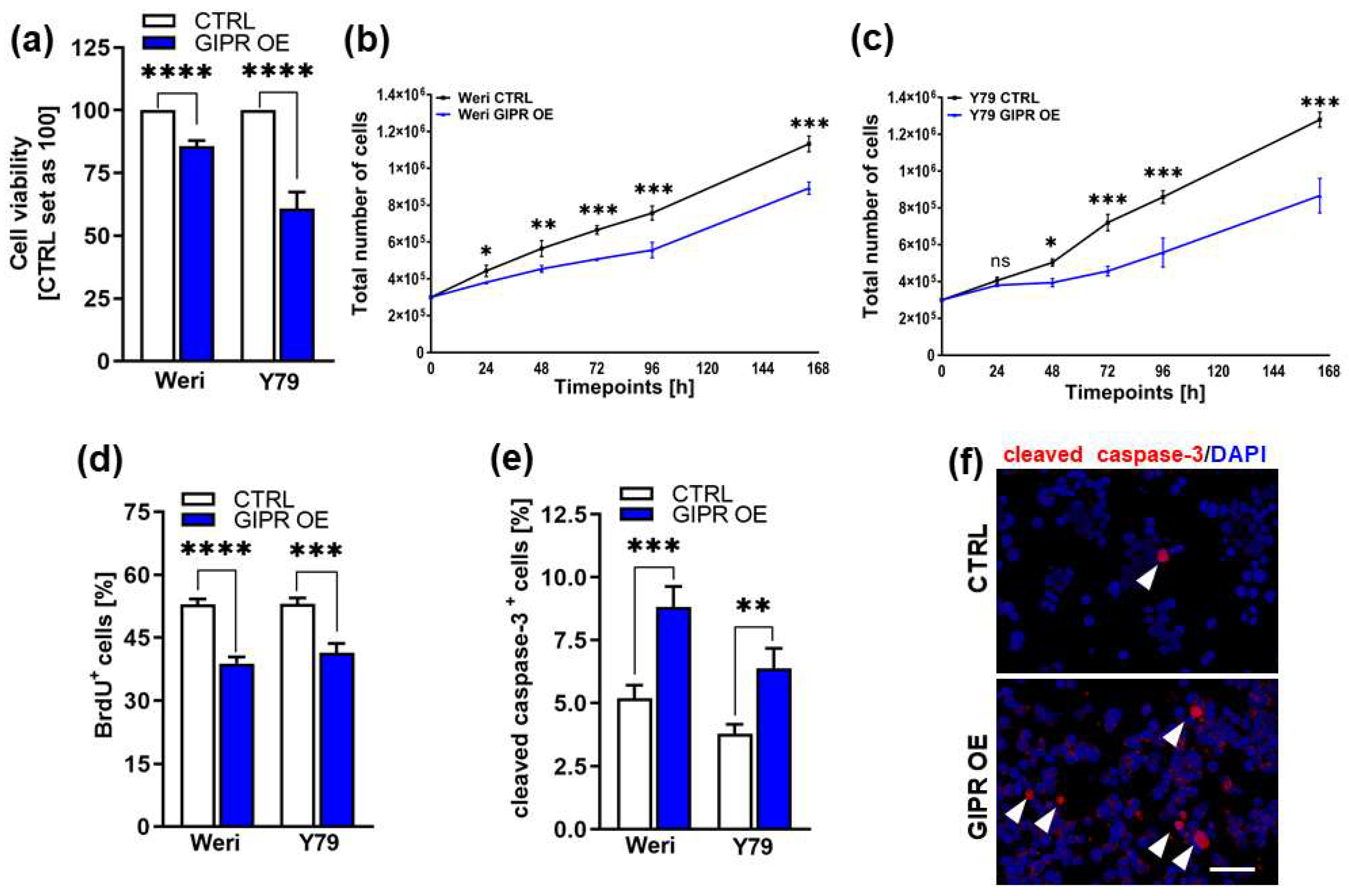
3.2. GIPR Overexpression Results in Decreased Cell Viability, Cell Growth, and Proliferation as Well as Increased Apoptosis in RB Cell Lines In Vitro
3.3. GIPR-Overexpressing RB Cells Form Significantly Smaller Tumors In Vivo
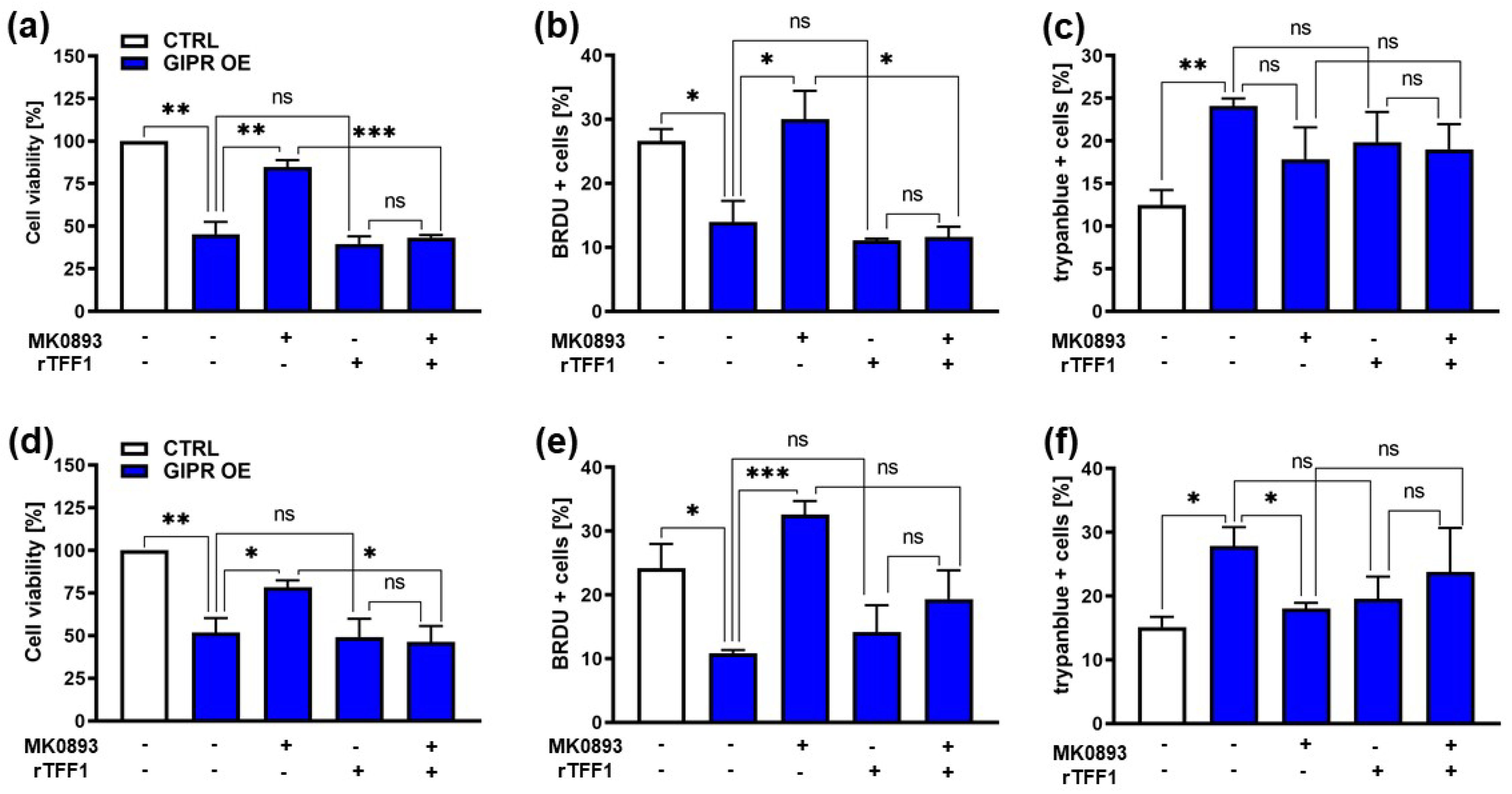
3.4. Impact of the Administration of a GIPR Inhibitor and/or Recombinant TFF1 on Cell Viability, Proliferation, and Cell Death of GIPR-Overexpressing RB Cell Lines
3.5. GIPR Expression in Retinoblastoma Cells and Its Regulation by miR-542-5p
3.6. GIPR Downstream Signaling Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Byroju, V.V.; Nadukkandy, A.S.; Cordani, M.; Kumar, L.D. Retinoblastoma: Present scenario and future challenges. Cell Commun. Signal. 2023, 21, 226. [Google Scholar] [CrossRef]
- Bouchoucha, Y.; Matet, A.; Berger, A.; Carcaboso, A.M.; Gerrish, A.; Moll, A.; Jenkinson, H.; Ketteler, P.; Dorsman, J.C.; Chantada, G.; et al. Retinoblastoma: From genes to patient care. Eur. J. Med. Genet. 2023, 66, 104674. [Google Scholar] [CrossRef] [PubMed]
- Bornfeld, N.; Lohmann, D.; Bechrakis, N.E.; Biewald, E. [Retinoblastoma]. Der Ophthalmol. 2020, 117, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kaliki, S. Retinoblastoma: A Major Review. Mymensingh Med. J. MMJ 2021, 30, 881–895. [Google Scholar] [PubMed]
- Dimaras, H.; Corson, T.W. Retinoblastoma, the visible CNS tumor: A review. J. Neurosci. Res. 2019, 97, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Rathore, S.; Verma, A.; Ratna, R.; Marwa, N.; Ghiya, Y.; Honavar, S.G.; Tiwari, A.; Das, S.; Varshney, A. Retinoblastoma: A review of the molecular basis of tumor development and its clinical correlation in shaping future targeted treatment strategies. Indian J. Ophthalmol. 2023, 71, 2662. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ottaviani, D.; Sefta, M.; Desbrousses, C.; Chapeaublanc, E.; Aschero, R.; Sirab, N.; Lubieniecki, F.; Lamas, G.; Tonon, L.; et al. A high-risk retinoblastoma subtype with stemness features, dedifferentiated cone states and neuronal/ganglion cell gene expression. Nat. Commun. 2021, 12, 5578. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Babst, N.; Schiemenz, C.; Schutz, J.S.; Heindl, L.M.; Ranjbar, M.; Kakkassery, V. A Narrative Review—Therapy Options and Therapy Failure in Retinoblastoma. Neurosignals 2022, 30, 39–58. [Google Scholar] [PubMed]
- Shields, C.L.; Bas, Z.; Laiton, A.; Silva, A.M.V.; Sheikh, A.; Lally, S.E.; Shields, J.A. Retinoblastoma: Emerging concepts in genetics, global disease burden, chemotherapy outcomes, and psychological impact. Eye 2023, 37, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Kaewkhaw, R.; Rojanaporn, D. Retinoblastoma: Etiology, Modeling, and Treatment. Cancers 2020, 12, 2304. [Google Scholar] [CrossRef]
- Munier, F.L.; Beck-Popovic, M.; Chantada, G.L.; Cobrinik, D.; Kivelä, T.T.; Lohmann, D.; Maeder, P.; Moll, A.C.; Carcaboso, A.M.; Moulin, A.; et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Prog. Retin. Eye Res. 2019, 73, 100764. [Google Scholar] [CrossRef] [PubMed]
- Manjandavida, F.P.; Shields, C.L. The role of intravitreal chemotherapy for retinoblastoma. Indian J. Ophthalmol. 2015, 63, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.H.; Abramson, D.H.; Ji, X.; Shields, C.L.; Teixeira, L.F.; Schefler, A.C.; Cassoux, N.; Hadjistilianou, D.; Berry, J.L.; Frenkel, S.; et al. Risk of Extraocular Extension in Eyes With Retinoblastoma Receiving Intravitreous Chemotherapy. JAMA Ophthalmol. 2017, 135, 1426. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Manjandavida, F.P.; Arepalli, S.; Kaliki, S.; Lally, S.E.; Shields, J.A. Intravitreal Melphalan for Persistent or Recurrent Retinoblastoma Vitreous Seeds: Preliminary Results. JAMA Ophthalmol. 2014, 132, 319. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Douglass, A.M.; Beggache, M.; Say, E.A.T.; Shields, J.A. Intravitreous Chemotherapy For Active Vitreous Seeding From Retinoblastoma. Retina 2016, 36, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Temming, P.; Arendt, M.; Viehmann, A.; Eisele, L.; Le Guin, C.H.D.; Schündeln, M.M.; Biewald, E.; Astrahantseff, K.; Wieland, R.; Bornfeld, N.; et al. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr. Blood Cancer 2017, 64, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.G.; Kuznetsoff, J.N.; Owens, D.A.; Gallo, R.A.; Kalahasty, K.; Cruz, A.M.; Kurtenbach, S.; Correa, Z.M.; Pelaez, D.; Harbour, J.W. Early Mechanisms of Chemoresistance in Retinoblastoma. Cancers 2022, 14, 4966. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Trefoil Factor Family (TFF) Peptides and Their Links to Inflammation: A Re-evaluation and New Medical Perspectives. Int. J. Mol. Sci. 2021, 22, 4909. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Trefoil Factor Family (TFF) Peptides and Their Diverse Molecular Functions in Mucus Barrier Protection and More: Changing the Paradigm. Int. J. Mol. Sci. 2020, 21, 4535. [Google Scholar] [CrossRef] [PubMed]
- Jahan, R.; Shah, A.; Kisling, S.G.; Macha, M.A.; Thayer, S.; Batra, S.K.; Kaur, S. Odyssey of trefoil factors in cancer: Diagnostic and therapeutic implications. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2020, 1873, 188362. [Google Scholar] [CrossRef]
- Busch, M.; Dünker, N. Trefoil factor family peptides—Friends or foes? Biomol. Concepts 2015, 6, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Große-Kreul, J.; Wirtz, J.J.; Beier, M.; Stephan, H.; Royer-Pokora, B.; Metz, K.; Dünker, N. Reduction of the tumorigenic potential of human retinoblastoma cell lines by TFF1 overexpression involves p53/caspase signaling and miR-18a regulation. Int. J. Cancer 2017, 141, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Metz, K.; Beier, M.; Biewald, E.; Dünker, N. Trefoil Factor Family 1 Expression Correlates with Clinical Outcome in Patients with Retinoblastoma. Retina 2018, 38, 2422. [Google Scholar] [CrossRef] [PubMed]
- Aschero, R.; Ganiewich, D.; Lamas, G.; Restrepo-Perdomo, C.A.; Ottaviani, D.; Zugbi, S.; Camarero, S.; Néspoli, E.; Vilanova, M.C.; Perez-Jaume, S.; et al. Immunohistochemical expression of TFF1 is a marker of poor prognosis in retinoblastoma. Pediatr. Blood Cancer 2024, 71, e30717. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.A.; Haase, A.; Miroschnikov, N.; Doege, A.; Biewald, E.; Bechrakis, N.E.; Beier, M.; Kanber, D.; Lohmann, D.; Metz, K.; et al. TFF1 in Aqueous Humor—A Potential New Biomarker for Retinoblastoma. Cancers 2022, 14, 677. [Google Scholar] [CrossRef]
- Busch, M.A.; Haase, A.; Alefeld, E.; Biewald, E.; Jabbarli, L.; Dünker, N. Trefoil Family Factor Peptide 1—A New Biomarker in Liquid Biopsies of Retinoblastoma under Therapy. Cancers 2023, 15, 4828. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Hayami, T.; Nakamura, K.; Kaisaki, P.J.; Someya, Y.; Wang, C.-Z.; Seino, S.; Seino, Y. Human Gastric Inhibitory Polypeptide Receptor: Cloning of the Gene (GIPR) and cDNA. Genomics 1995, 29, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Gremlich, S.; Porret, A.; Hani, E.H.; Cherif, D.; Vionnet, N.; Froguel, P.; Thorens, B. Cloning, functional expression, and chromosomal localization of the human pancreatic islet glucose-dependent insulinotropic polypeptide receptor. Diabetes 1995, 44, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Göke, R.; Lankat-Buttgereit, B.; Fehmann, H.-C.; Bode, H.P.; Göke, B. Molecular cloning, functional expression, and signal transduction of the GIP-receptor cloned from a human insulinoma. FEBS Lett. 1995, 373, 23–29. [Google Scholar] [CrossRef]
- Dubeykovskaya, Z.; Dubeykovskiy, A.; Solal-Cohen, J.; Wang, T.C. Secreted Trefoil Factor 2 Activates the CXCR4 Receptor in Epithelial and Lymphocytic Cancer Cell Lines. J. Biol. Chem. 2009, 284, 3650–3662. [Google Scholar] [CrossRef]
- Dieckow, J.; Brandt, W.; Hattermann, K.; Schob, S.; Schulze, U.; Mentlein, R.; Ackermann, P.; Sel, S.; Paulsen, F.P. CXCR4 and CXCR7 Mediate TFF3-Induced Cell Migration Independently From the ERK1/2 Signaling Pathway. Investig. Ophthalmol. Vis. Sci. 2016, 57, 56. [Google Scholar] [CrossRef] [PubMed]
- Dupre, J.; Ross, S.A.; Watson, D.; Brown, J.C. Stimulation Of Insulin Secretion By Gastric Inhibitory Polypeptide In Man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Regazzo, D.; Barbot, M.; Scaroni, C.; Albiger, N.; Occhi, G. The pathogenic role of the GIP/GIPR axis in human endocrine tumors: Emerging clinical mechanisms beyond diabetes. Rev. Endocr. Metab. Disord. 2020, 21, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Regazzo, D.; Bertazza, L.; Galletta, E.; Barollo, S.; Mondin, A.; Zovato, S.; Iacobone, M.; Zilio, E.; Scaroni, C.; Radu, C.M.; et al. The GIP/GIPR axis in medullary thyroid cancer: Clinical and molecular findings. Endocr.-Relat. Cancer 2022, 29, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Usdin, T.B.; Mezey, V.; Button, C.; Brownstein, J.; Bonner, T.I. Gastric Inhibitory Polypeptide Receptor, a Member of the Secretin-Vasoactive Intestinal Peptide Receptor Family, Is Widely Distributed in Peripheral Organs and the Brain. Endocrinology 1993, 133, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.N.; Basu, D.; Skoblenick, K.J.; Castellano, J.M.; Pontoriero, G.; Thomas, N. Mapping Of Human Brain For Glucose-Dependent Insulinotropic Polypeptide (Gip) And Gip Receptors Expression: Implications In Schizophrenia. Schizophr. Res. 2010, 2–3, 528. [Google Scholar] [CrossRef]
- Verma, M.K.; Goel, R.; Krishnadas, N.; Nemmani, K.V.S. Targeting glucose-dependent insulinotropic polypeptide receptor for neurodegenerative disorders. Expert Opin. Ther. Targets 2018, 22, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Samms, R.J.; Sloop, K.W.; Gribble, F.M.; Reimann, F.; Adriaenssens, A.E. GIPR Function in the Central Nervous System: Implications and Novel Perspectives for GIP-Based Therapies in Treating Metabolic Disorders. Diabetes 2021, 70, 1938–1944. [Google Scholar] [CrossRef]
- Waser, B.; Rehmann, R.; Sanchez, C.; Fourmy, D.; Reubi, J.C. Glucose-Dependent Insulinotropic Polypeptide Receptors in Most Gastroenteropancreatic and Bronchial Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2012, 97, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Körner, M.; Waser, B.; Reubi, J.C. Does Somatostatin or Gastric Inhibitory Peptide Receptor Expression Correlate with Tumor Grade and Stage in Gut Neuroendocrine Tumors? Neuroendocrinology 2015, 101, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Waser, B.; Beetschen, K.; Pellegata, N.S.; Reubi, J.C. Incretin Receptors in Non-Neoplastic and Neoplastic Thyroid C Cells in Rodents and Humans: Relevance for Incretin-Based Diabetes Therapy. Neuroendocrinology 2011, 94, 291–301. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Ao, Z.; Kim, S.-J.; Warnock, G.; McIntosh, C.H. Suppression of p38 MAPK and JNK via Akt-mediated Inhibition of Apoptosis Signal-regulating Kinase 1 Constitutes a Core Component of the β-Cell Pro-survival Effects of Glucose-dependent Insulinotropic Polypeptide. J. Biol. Chem. 2009, 284, 30372–30382. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, D.; Wang, B.; Feuerstein, J.D.; Sinclair, J.A.; Bijpuria, P.; Jepeal, L.I.; Wolfe, M.M. Glucose-dependent insulinotropic polypeptide stimulates the proliferation of colorectal cancer cells. Regul. Pept. 2010, 163, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Yabe, D.; Seino, Y. Two incretin hormones GLP-1 and GIP: Comparison of their actions in insulin secretion and β cell preservation. Prog. Biophys. Mol. Biol. 2011, 107, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Islet α cells and glucagon—Critical regulators of energy homeostasis. Nat. Rev. Endocrinol. 2015, 11, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Pospisilik, J.A.; Martin, J.; Doty, T.; Ehses, J.A.; Pamir, N.; Lynn, F.C.; Piteau, S.; Demuth, H.-U.; McIntosh, C.H.S.; Pederson, R.A. Dipeptidyl peptidase IV inhibitor treatment stimulates β-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 2003, 52, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Casilla, V.R.; Doty, T.; Pospisilik, J.A.; Winter, K.D.; Demuth, H.-U.; Pederson, R.A.; McIntosh, C.H.S. Glucose-Dependent Insulinotropic Polypeptide Promotes β-(INS-1) Cell Survival via Cyclic Adenosine Monophosphate-Mediated Caspase-3 Inhibition and Regulation of p38 Mitogen-Activated Protein Kinase. Endocrinology 2003, 144, 4433–4445. [Google Scholar] [CrossRef] [PubMed]
- McFall, R.C.; Sery, T.W.; Makadon, M. Characterization of a New Continuous Cell Line Derived from a Human Retinoblastoma. Cancer Res. 1977, 37, 1003–1010. [Google Scholar] [PubMed]
- Reid, T.W.; Albert, D.M.; Rabson, A.S.; Russell, P.; Craft, J.; Chu, E.W.; Tralka, T.S.; Wilcox, J.L. Characteristics of an Established Cell Line of Retinoblastoma2. JNCI J. Natl. Cancer Inst. 1974, 53, 347–360. [Google Scholar] [CrossRef]
- Busch, M.; Philippeit, C.; Weise, A.; Dünker, N. Re-characterization of established human retinoblastoma cell lines. Histochem. Cell Biol. 2015, 143, 325–338. [Google Scholar] [CrossRef]
- Campeau, E.; Ruhl, V.E.; Rodier, F.; Smith, C.L.; Rahmberg, B.L.; Fuss, J.O.; Campisi, J.; Yaswen, P.; Cooper, P.K.; Kaufman, P.D. A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. PLoS ONE 2009, 4, e6529. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.; Motsch, N.; Zhu, J.Y.; Barth, S.; Okoniewski, M.; Reineke, T.; Tinguely, M.; Faggioni, A.; Trivedi, P.; Meister, G.; et al. microRNA profiling in Epstein–Barr virus-associated B-cell lymphoma. Nucleic Acids Res. 2011, 39, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Heinkelein, M.; Heldmann, M.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. Foamy Virus Capsids Require the Cognate Envelope Protein for Particle Export. J. Virol. 1999, 73, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Schwartz, J.P.; Tanaka, K.; Brady, R.O.; Reiser, J. High-Titer Human Immunodeficiency Virus Type 1-Based Vector Systems for Gene Delivery into Nondividing Cells. J. Virol. 1998, 72, 8873–8883. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, A.; Mellor, R.; Panzarella, G.; Aimes, R.T.; Hooper, J.D.; Marchenko, N.D.; Quigley, J.P. A Quantitative Analysis of Rate-limiting Steps in the Metastatic Cascade Using Human-specific Real-Time Polymerase Chain Reaction1. Cancer Res. 2002, 62, 7083–7092. [Google Scholar] [PubMed]
- Palmer, T.D.; Lewis, J.; Zijlstra, A. Quantitative Analysis of Cancer Metastasis using an Avian Embryo Model. JoVE (J. Vis. Exp.) 2011, 51, e2815. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Papior, D.; Stephan, H.; Dünker, N. Characterization of etoposide- and cisplatin-chemoresistant retinoblastoma cell lines. Oncol. Rep. 2018, 39, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Klein, S.; Große-Kreul, J.; Scheiner, O.; Metz, K.; Stephan, H.; Dünker, N. p53, miR-34a and EMP1—Newly Identified Targets of TFF3 Signaling in Y79 Retinoblastoma Cells. Int. J. Mol. Sci. 2019, 20, 4129. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- He, R.; Li, X.; Liang, L.; Xie, Y.; Luo, D.; Ma, J.; Peng, Z.; Hu, X.; Chen, G. The suppressive role of miR-542-5p in NSCLC: The evidence from clinical data and in vivo validation using a chick chorioallantoic membrane model. BMC Cancer 2017, 17, 655. [Google Scholar] [CrossRef] [PubMed]
- Philippeit, C.; Busch, M.; Dünker, N. Epigenetic Control of Trefoil Factor Family (TFF) Peptide Expression in Human Retinoblastoma Cell Lines. Cell. Physiol. Biochem. 2014, 34, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-G.; Li, Y.-F.; Pan, B.-L.; Wang, L.-P.; Zhang, Y.; Lee, W.-H.; Zhang, Y. Trefoil factor 1 gene alternations and expression in colorectal carcinomas. Tumori J. 2013, 99, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Oh, R.R.; Park, J.Y.; Lee, J.H.; Shin, M.S.; Kim, H.S.; Lee, H.K.; Kim, Y.S.; Kim, S.Y.; Lee, S.H.; et al. Somatic mutations of the trefoil factor family 1 gene in gastric cancer. Gastroenterology 2000, 119, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Shekarriz, R.; Kochaki, N.; Eslami-Jouibari, M.; Omrani-Nava, V.; Ahmadi, M.; Alizadeh-Navaei, R. TFF1 gene single nucleotide polymorphism (rs3761376) and colorectal cancer risk. Mol. Biol. Rep. 2022, 49, 10127–10131. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Z.; Wang, J.; Du, M.; Li, B.; Zhang, L.; Li, Q.; Xu, J.; Wang, L.; Li, F.; et al. A functional polymorphism in TFF1 promoter is associated with the risk and prognosis of gastric cancer. Int. J. Cancer 2018, 142, 1805–1816. [Google Scholar] [CrossRef]
- Yio, X.; Diamond, M.; Zhang, J.-Y.; Weinstein, H.; Wang, L.-H.; Werther, L.; Itzkowitz, S. Trefoil Factor Family-1 Mutations Enhance Gastric Cancer Cell Invasion Through Distinct Signaling Pathways. Gastroenterology 2006, 130, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Fourmy, D.; Cordomi, A.; Tikhonova, I.G.; Gigoux, V. GIP receptor: Expression in neuroendocrine tumours, internalization, signalling from endosomes and structure-function relationship studies. Peptides 2020, 125, 170229. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.K.; Maxwell, J.E.; Carr, J.C.; Wang, D.; O’Dorisio, M.S.; O’Dorisio, T.M.; Howe, J.R. GIPR expression in gastric and duodenal neuroendocrine tumors. J. Surg. Res. 2014, 190, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Karpathakis, A.; Dibra, H.; Pipinikas, C.; Feber, A.; Morris, T.; Francis, J.; Oukrif, D.; Mandair, D.; Pericleous, M.; Mohmaduvesh, M.; et al. Prognostic Impact of Novel Molecular Subtypes of Small Intestinal Neuroendocrine Tumor. Clin. Cancer Res. 2016, 22, 250–258. [Google Scholar] [CrossRef]
- Costa, M.H.S.; Latronico, A.C.; Martin, R.M.; Barbosa, A.S.; Almeida, M.Q.; Lotfi, C.F.P.; Valassi, H.P.L.; Nishi, M.Y.; Lucon, A.M.; Siqueira, S.A.; et al. Expression profiles of the glucose-dependent insulinotropic peptide receptor and LHCGR in sporadic adrenocortical tumors. J. Endocrinol. 2009, 200, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hu, C.; Zhen, Y.; Pang, B.; Yi, H.; Chen, X. Pristimerin inhibits glioma progression by targeting AGO2 and PTPN1 expression via miR-542-5p. Biosci. Rep. 2019, 39, BSR20182389. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Dantas Machado, A.C.; Ding, Y.; Chen, Z.; Qin, P.Z.; Rohs, R.; Chen, L. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013, 41, 8368–8376. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Qu, L.; Dai, S.; Li, Y.; Wang, H.; Feng, Y.; Chen, X.; Jiang, L.; Guo, M.; Li, J.; et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 2021, 12, 2280. [Google Scholar] [CrossRef] [PubMed]
- Kondapuram, S.K.; Ramachandran, H.K.; Arya, H.; Coumar, M.S. Targeting survivin for cancer therapy: Strategies, small molecule inhibitors and vaccine based therapeutics in development. Life Sci. 2023, 335, 122260. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Pascual, A.; Mitchell, K.; Siebzehnrubl, F.A.; Lathia, J.D. FGF2: A novel druggable target for glioblastoma? Expert Opin. Ther. Targets 2020, 24, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Chandarana, R.; D’Souza, J.S.; Coutinho, E.C. Glucose-Dependent Insulinotropic Polypeptide Receptor (GIPR). In Encyclopedia of Signaling Molecules; Springer: Berlin/Heidelberg, Germany, 2012; pp. 773–779. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | 5′-3′ Sequence |
|---|---|
| ENO2 FW | CTACCACCGTCTGAGTCTGC |
| ENO2 RV | CCTTCAGGACACCTTTGCCT |
| ERBB2 FW | GTTCCCGGATTTTTGTGGGC |
| ERBB2 RV | GTGGTACTTCAATTGCGACTCA |
| p27 FW | CTGCAACCGACGATTCTTCT |
| p27 RV | GCATTTGGGGAACCGTCTGA |
| p53 FW | TGTGACTTGCACGTACTCCC |
| p53 RV | ACCATCGCTATCTGAGCAGC |
| Survivin FW | TGAGAACGAGCCAGACTTGG |
| Survivin RV | TGGTTTCCTTTGCATGGGGT |
| FGFb FW | CCGTTACCTGGCTATGAAGG |
| FGFb RV | AAAGAAACACTCATCCGTAACACA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haase, A.; Alefeld, E.; Yalinci, F.; Meenen, D.V.; Busch, M.A.; Dünker, N. Gastric Inhibitory Polypeptide Receptor (GIPR) Overexpression Reduces the Tumorigenic Potential of Retinoblastoma Cells. Cancers 2024, 16, 1656. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16091656
Haase A, Alefeld E, Yalinci F, Meenen DV, Busch MA, Dünker N. Gastric Inhibitory Polypeptide Receptor (GIPR) Overexpression Reduces the Tumorigenic Potential of Retinoblastoma Cells. Cancers. 2024; 16(9):1656. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16091656
Chicago/Turabian StyleHaase, André, Emily Alefeld, Fatma Yalinci, Dario Van Meenen, Maike Anna Busch, and Nicole Dünker. 2024. "Gastric Inhibitory Polypeptide Receptor (GIPR) Overexpression Reduces the Tumorigenic Potential of Retinoblastoma Cells" Cancers 16, no. 9: 1656. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16091656