
Naphthalene Monoimides with Peri-Annulated Disulfide Bridge—Synthesis and Electrochemical Redox Activity
 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. Synthesis of (N-octyl)-3,4,5,6-tetrahalo-1,8-naphthalimides 3 and 4
Synthesis of (N-octyl)-3,4,5,6-tetrachloro-1,8-naphthalimide 3
Synthesis of (N-octyl)-3,4,5,6-tetrabromo-1,8-naphthalimide 4
2.1.2. Synthesis of (N-butyl)-3,4,5,6-tetrahalo-1,8-naphthalimides 5 and 6
Synthesis of (N-butyl)-3,4,5,6-tetrachloro-1,8-naphthalimide 5
Synthesis of (N-butyl)-3,4,5,6-tetrabromo-1,8-naphthalimide 6
2.1.3. Synthesis of 6-Alkyl-3,9-dihalo-5H-[1,2]dithiolo[3′,4′,5′:4,5]naphtho[1,8-cd]pyridine-5,7(6H)-diones 7–10
Synthesis of 3,9-Dichloro-6-octyl-5H-[1,2]dithiolo[3′,4′,5′:4,5]naphtho[1,8-cd]-pyridine-5,7(6H)-dione SCl8
Synthesis of 3,9-Dibromo-6-octyl-5H-[1,2]dithiolo[3′,4′,5′:4,5]naphtho[1,8-cd]-pyridine-5,7(6H)-dione SBr8
Synthesis of 6-Butyl-3,9-dichloro-5H-[1,2]dithiolo[3′,4′,5′:4,5]naphtho[1,8-cd]-pyridine-5,7(6H)-dione SCl4
Synthesis of 3,9-Dibromo-6-butyl-5H-[1,2]dithiolo[3′,4′,5′:4,5]naphtho[1,8-cd]-pyridine-5,7(6H)-dione SBr4
2.2. Electrochemical Characterization
3. Results and Discussion
3.1. Design Concept
3.2. Synthesis
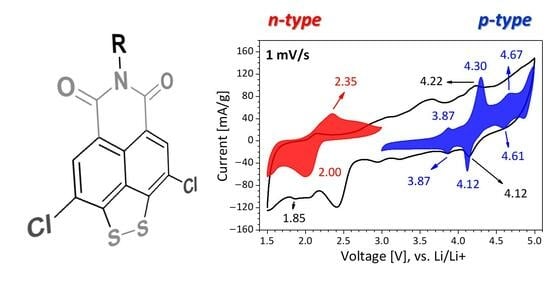
3.3. Redox Properties of Peri-Substituted Dichalchogenides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Kim, T.; Song, W.; Son, D.-Y.; Ono, L.K.; Qi, Y. Lithium-ion batteries: Outlook on present, future, and hybridized technologies. J. Mater. Chem. A 2019, 7, 2942–2964. [Google Scholar] [CrossRef]
- Kim, H.-J.; Krishna, T.; Zeb, K.; Rajangam, V.; Gopi, C.V.V.M.; Sambasivam, S.; Raghavendra, K.V.G.; Obaidat, I.M. A Comprehensive Review of Li-Ion Battery Materials and Their Recycling Techniques. Electronics 2020, 9, 1161. [Google Scholar] [CrossRef]
- Deng, C.; Li, X.; Chen, R.; Ye, K.; Lipton, J.; Maclean, S.A.; Wang, H.; Taylor, A.D.; Weng, G.M. Recent advances in rocking chair batteries and beyond. Energy Storage Mater. 2023, 60, 102820. [Google Scholar] [CrossRef]
- Sironval, V.; Scagliarini, V.; Murugadoss, S.; Tomatis, M.; Yakoub, Y.; Turci, F.; Hoet, P.; Lison, D.; van den Brule, S. LiCoO2 particles used in Li-ion batteries induce primary mutagenicity in lung cells via their capacity to generate hydroxyl radicals. Part. Fibre Toxicol. 2020, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, R.; Chordia, M.; Nordelöf, A. Quantifying the life-cycle health impacts of a cobalt-containing lithium-ion battery. Int. J. Life Cycle Assess. 2022, 27, 1106–1118. [Google Scholar] [CrossRef]
- Wentker, M.; Greenwood, M.; Leker, J. A Bottom-Up Approach to Lithium-Ion Battery Cost Modeling with a Focus on Cathode Active Materials. Energies 2019, 12, 504. [Google Scholar] [CrossRef]
- Mrozik, W.; Rajaeifar, M.A.; Heidrich, O.; Christensen, P. Environmental impacts, pollution sources and pathways of spent lithium-ion batteries. Energy Environ. Sci. 2021, 14, 6099–6121. [Google Scholar] [CrossRef]
- Li, L.; Yin, Y.; Hei, J.; Wan, X.; Li, M.; Cui, Y. Molecular Engineering of Aromatic Imides for Organic Secondary Batteries. Small 2021, 17, 2005752. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, H.; Fan, K.; Zhang, G.; Tang, M.; Gao, Y.; Zhang, C.; Guan, L.; Mao, M.; Liu, H.; et al. A Recyclable and Scalable High-Capacity Organic Battery. Angew. Chem. Int. Ed. 2023, 62, e202302539. [Google Scholar] [CrossRef]
- Guo, W.; Fu, Y. Electrochemistry of Electrode Materials Containing S−Se Bonds for Rechargeable Batteries. Chem.A Eur. J. 2020, 26, 13322–13331. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Q.; Li, L.; Niu, Z.; Chen, J. Design Strategies toward Enhancing the Performance of Organic Electrode Materials in Metal-Ion Batteries. Chem 2018, 4, 2786–2813. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Guo, W.; Fu, Y. Organosulfides: An Emerging Class of Cathode Materials for Rechargeable Lithium Batteries. Acc. Chem. Res. 2019, 52, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Shadike, Z.; Tan, S.; Wang, Q.-C.; Lin, R.; Hu, E.; Qu, D.; Yang, X.-Q. Review on organosulfur materials for rechargeable lithium batteries. Mater. Horizons 2021, 8, 471–500. [Google Scholar] [CrossRef]
- Visco, S.J.; Liu, M.; De Jonghe, L.C. Ambient Temperature High-Rate Lithium/Organosulfur Batteries. J. Electrochem. Soc. 1990, 137, 1191–1192. [Google Scholar] [CrossRef]
- Li, F.; Si, Y.; Liu, B.; Li, Z.; Fu, Y. Lithium Benzenedithiolate Catholytes for Rechargeable Lithium Batteries. Adv. Funct. Mater. 2019, 29, 1902223. [Google Scholar] [CrossRef]
- Bhargav, A.; Patil, S.V.; Fu, Y. A phenyl disulfide@CNT composite cathode for rechargeable lithium batteries. Sustain. Energy Fuels 2017, 1, 1007–1012. [Google Scholar] [CrossRef]
- Wang, D.-Y.Y.; Si, Y.; Li, J.; Fu, Y. Tuning the electrochemical behavior of organodisulfides in rechargeable lithium batteries using N-containing heterocycles. J. Mater. Chem. A 2019, 7, 7423–7429. [Google Scholar] [CrossRef]
- Sakai, N.; Mareda, J.; Vauthey, E.; Matile, S. Core-substituted naphthalenediimides. Chem. Commun. 2010, 46, 4225. [Google Scholar] [CrossRef]
- Chen, S.; Jia, T.; Zhou, G.; Zhang, C.; Hou, Q.; Wang, Y.; Luo, S.; Shi, G.; Zeng, Y. A Cross-Linked Triphenylamine-Based Polymer Cathode Material with Dual Anion-Cation Reversible Insertion for Lithium Ion Battery. J. Electrochem. Soc. 2019, 166, A2543–A2548. [Google Scholar] [CrossRef]
- Al Kobaisi, M.; Bhosale, S.V.; Latham, K.; Raynor, A.M.; Bhosale, S.V. Functional Naphthalene Diimides: Synthesis, Properties, and Applications. Chem. Rev. 2016, 116, 11685–11796. [Google Scholar] [CrossRef]
- Zagranyarski, Y.; Mutovska, M.; Petrova, P.; Tomova, R.; Ivanov, P.; Stoyanov, S. Dioxin-annulated 1,8-naphthalimides—Synthesis, spectral and electrochemical properties, and application in OLED. Dye. Pigment. 2021, 184, 108585. [Google Scholar] [CrossRef]
- Said, A.I.; Staneva, D.; Angelova, S.; Grabchev, I. Self-Associated 1,8-Naphthalimide as a Selective Fluorescent Chemosensor for Detection of High pH in Aqueous Solutions and Their Hg2+ Contamination. Sensors 2023, 23, 399. [Google Scholar] [CrossRef]
- Banerjee, S.; Veale, E.B.; Phelan, C.M.; Murphy, S.A.; Tocci, G.M.; Gillespie, L.J.; Frimannsson, D.O.; Kelly, J.M.; Gunnlaugsson, T. Recent advances in the development of 1,8-naphthalimide based DNA targeting binders, anticancer and fluorescent cellular imaging agents. Chem. Soc. Rev. 2013, 42, 1601–1618. [Google Scholar] [CrossRef]
- Lee, S.; Hong, J.; Jung, S.K.; Ku, K.; Kwon, G.; Seong, W.M.; Kim, H.; Yoon, G.; Kang, I.; Hong, K.; et al. Charge-transfer complexes for high-power organic rechargeable batteries. Energy Storage Mater. 2019, 20, 462–469. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Zagranyarski, Y.; Müller, C.; Schulz, M.; Kupfer, S.; Görls, H.; El-khateeb, M.; Gräfe, S.; Dietzek, B.; Peneva, K.; et al. [FeFe]-Hydrogenase H-cluster mimics mediated by naphthalene monoimide derivatives of peri-substituted dichalcogenides. Dalt. Trans. 2017, 46, 11180–11191. [Google Scholar] [CrossRef] [PubMed]
- Inamasu, T.; Yoshitoku, D.; Sumi-otorii, Y.; Tani, H.; Ono, N. Electrochemical Behaviors of Naphtho[1,8-cd][1,2]dithiol, Dibenzo[c,e][1,2]dithiin, and Naphtho[1,8-cd:4,5-cʹdʹ]bis[1,2]dithiol. J. Electrochem. Soc. 2003, 150, A128. [Google Scholar] [CrossRef]
- Liang, Y.; Tao, Z.; Chen, J. Organic electrode materials for rechargeable lithium batteries. Adv. Energy Mater. 2012, 2, 742–769. [Google Scholar] [CrossRef]
- Mauger, A.; Julien, C.; Paolella, A.; Armand, M.; Zaghib, K. Recent Progress on Organic Electrodes Materials for Rechargeable Batteries and Supercapacitors. Materials 2019, 12, 1770. [Google Scholar] [CrossRef]
- Yin, X.; Sarkar, S.; Shi, S.; Huang, Q.; Zhao, H.; Yan, L.; Zhao, Y.; Zhang, J. Recent Progress in Advanced Organic Electrode Materials for Sodium-Ion Batteries: Synthesis, Mechanisms, Challenges and Perspectives. Adv. Funct. Mater. 2020, 30, 1908445. [Google Scholar] [CrossRef]
- Mutovska, M.; Skabeev, A.; Konstantinov, K.; Cabanetos, C.; Stoyanov, S.; Zagranyarski, Y. One-pot synthesis of fused-rings heterocyclic systems based on symmetrically benzofuran annulated 1,8-naphthalimides. Dye. Pigment. 2023, 220, 4–10. [Google Scholar] [CrossRef]
- Seybold, G. New perylene and violanthrone dyestuffs for fluorescent collectors. Dye. Pigment. 1989, 11, 303–317. [Google Scholar] [CrossRef]
- Langhals, H. Cyclic Carboxylic Imide Structures as Structure Elements of High Stability. Novel Developments in Perylene Dye Chemistry. Heterocycles 1995, 40, 477. [Google Scholar] [CrossRef]
- Karuppasamy, K.; Theerthagiri, J.; Vikraman, D.; Yim, C.-J.; Hussain, S.; Sharma, R.; Maiyalagan, T.; Qin, J.; Kim, H.-S. Ionic Liquid-Based Electrolytes for Energy Storage Devices: A Brief Review on Their Limits and Applications. Polymers 2020, 12, 918. [Google Scholar] [CrossRef]
- Guo, W.; Wang, D.; Chen, Q.; Fu, Y. Advances of Organosulfur Materials for Rechargeable Metal Batteries. Adv. Sci. 2022, 9, 2103989. [Google Scholar] [CrossRef] [PubMed]
- Vadehra, G.S.; Maloney, R.P.; Garcia-Garibay, M.A.; Dunn, B. Naphthalene Diimide Based Materials with Adjustable Redox Potentials: Evaluation for Organic Lithium-Ion Batteries. Chem. Mater. 2014, 26, 7151–7157. [Google Scholar] [CrossRef]
- Kye, H.; Kang, Y.; Jang, D.; Kwon, J.E.; Kim, B.-G. p-Type Redox-Active Organic Electrode Materials for Next-Generation Rechargeable Batteries. Adv. Energy Sustain. Res. 2022, 3, 2200030. [Google Scholar] [CrossRef]
- Speer, M.E.; Kolek, M.; Jassoy, J.J.; Heine, J.; Winter, M.; Bieker, P.M.; Esser, B. Thianthrene-functionalized polynorbornenes as high-voltage materials for organic cathode-based dual-ion batteries. Chem. Commun. 2015, 51, 15261–15264. [Google Scholar] [CrossRef] [PubMed]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2000; ISBN 978-0-471-04372-0. [Google Scholar]
- Acker, P.; Rzesny, L.; Marchiori, C.F.N.; Araujo, C.M.; Esser, B. π-Conjugation Enables Ultra-High Rate Capabilities and Cycling Stabilities in Phenothiazine Copolymers as Cathode-Active Battery Materials. Adv. Funct. Mater. 2019, 29, 1906436. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutovska, M.; Simeonova, N.; Stoyanov, S.; Zagranyarski, Y.; Stanchovska, S.; Marinova, D. Naphthalene Monoimides with Peri-Annulated Disulfide Bridge—Synthesis and Electrochemical Redox Activity. Materials 2023, 16, 7471. https://0-doi-org.brum.beds.ac.uk/10.3390/ma16237471
Mutovska M, Simeonova N, Stoyanov S, Zagranyarski Y, Stanchovska S, Marinova D. Naphthalene Monoimides with Peri-Annulated Disulfide Bridge—Synthesis and Electrochemical Redox Activity. Materials. 2023; 16(23):7471. https://0-doi-org.brum.beds.ac.uk/10.3390/ma16237471
Chicago/Turabian StyleMutovska, Monika, Natali Simeonova, Stanimir Stoyanov, Yulian Zagranyarski, Silva Stanchovska, and Delyana Marinova. 2023. "Naphthalene Monoimides with Peri-Annulated Disulfide Bridge—Synthesis and Electrochemical Redox Activity" Materials 16, no. 23: 7471. https://0-doi-org.brum.beds.ac.uk/10.3390/ma16237471