
Unveiling the Role of Tryptophan 2,3-Dioxygenase in the Angiogenic Process

 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TDO and IDO1 Expression in HUVECs and ECFCs
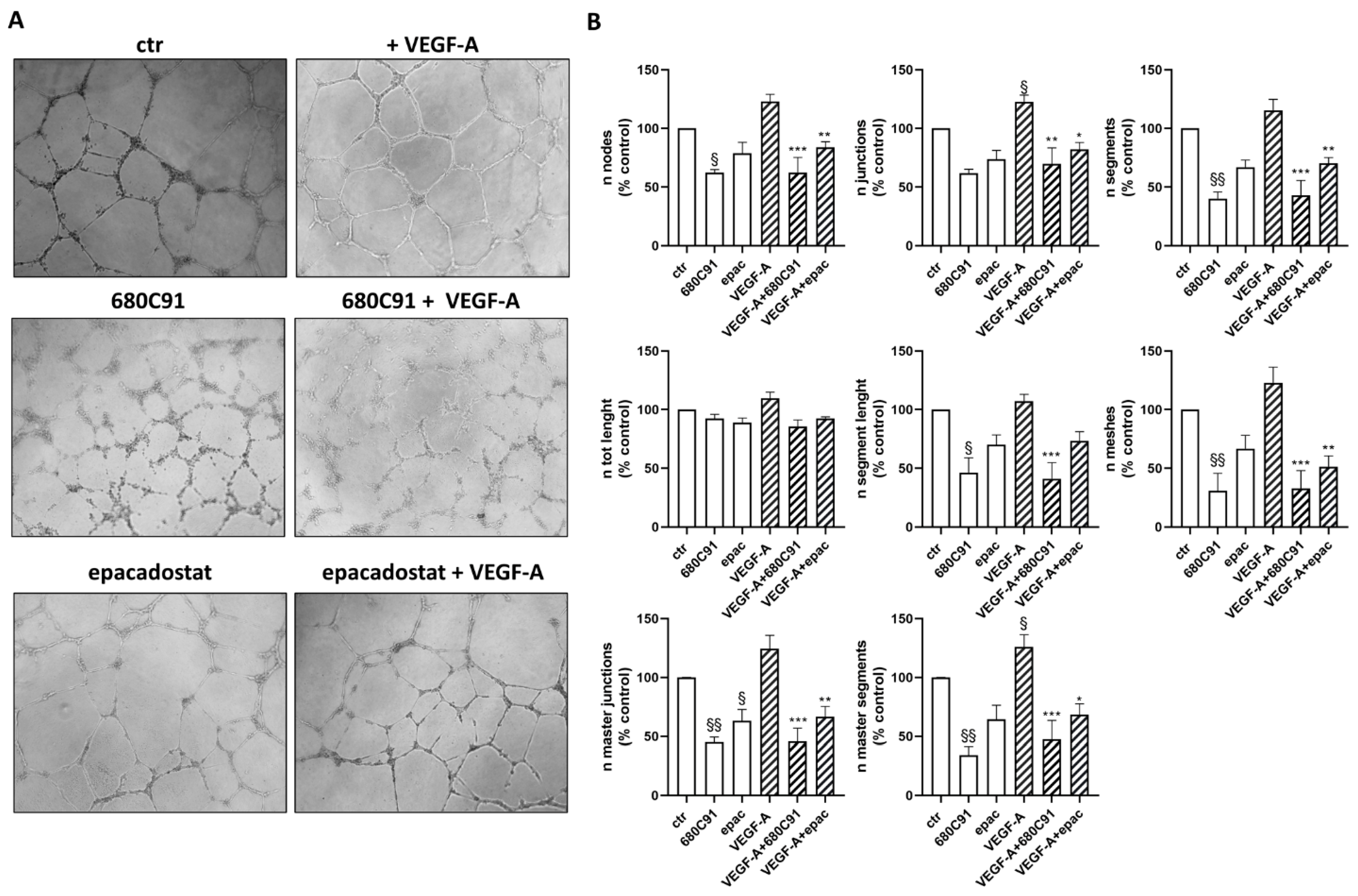
2.2. Effect of TDO and IDO1 Inhibition on In Vitro Capillary-like Structures
2.3. TDO but Not IDO1 Is Involved in HUVEC Proliferation
2.4. Endothelial Signaling Linked to TDO Activation
2.5. Role of TDO on A375-HUVEC Communication
2.6. MMP Release in the Co-Cultures
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Co-Cultures
4.3. Real-Time PCR
4.4. Sanger Sequencing
4.5. Immunofluorescence
4.6. Cell Proliferation
4.7. In Vitro Capillary Morphogenesis
4.8. ELISA Assay for Kynurenine Determination
4.9. Western Blot Analysis
4.10. Gelatin Zymography
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Badawy, A.A.B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed]
- Schramme, F.; Crosignani, S.; Frederix, K.; Hoffmann, D.; Pilotte, L.; Stroobant, V.; Preillon, J.; Driessens, G.; van den Eynde, B.J. Inhibition of Tryptophan-Dioxygenase Activity Increases the Antitumor Efficacy of Immune Checkpoint Inhibitors. Cancer Immunol. Res. 2020, 8, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Oweira, H.; Lahdou, I.; Mehrle, S.; Khajeh, E.; Nikbakhsh, R.; Ghamarnejad, O.; Terness, P.; Reißfelder, C.; Sadeghi, M.; Ramouz, A. Kynurenine Is the Main Metabolite of Tryptophan Degradation by Tryptophan 2,3-Dioxygenase in HepaRG Tumor Cells. J. Clin. Med. 2023, 12, 3177. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Peng, L.; Tao, B.; Yin, S.; Lyu, L.; Ding, H.; Yang, X. TDO2 and Tryptophan Metabolites Promote Kynurenine/AhR Signals to Facilitate Glioma Progression and Immunosuppression. Am. J. Cancer Res. 2022, 12, 2558–2575. [Google Scholar] [PubMed]
- Cecchi, M.; Paccosi, S.; Silvano, A.; Eid, A.H.; Parenti, A. Dexamethasone Induces the Expression and Function of Tryptophan-2-3-Dioxygenase in SK-MEL-28 Melanoma Cells. Pharmaceuticals 2021, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, M.; Mannini, A.; Lapucci, A.; Silvano, A.; Lulli, M.; Luceri, C.; D’Ambrosio, M.; Chiarugi, A.; Eid, A.H.; Parenti, A. Dexamethasone Promotes a Stem-Like Phenotype in Human Melanoma Cells via Tryptophan 2,3 Dioxygenase. Front. Pharmacol. 2022, 13, 911019. [Google Scholar] [CrossRef]
- Dewing, D.; Emmett, M.; Pritchard Jones, R. The Roles of Angiogenesis in Malignant Melanoma: Trends in Basic Science Research over the Last 100 Years. ISRN Oncol. 2012, 2012, 546927. [Google Scholar] [CrossRef]
- Pandita, A.; Ekstrand, M.; Bjursten, S.; Zhao, Z.; Fogelstrand, P.; Le Gal, K.; Ny, L.; Bergo, M.O.; Karlsson, J.; Nilsson, J.A.; et al. Intussusceptive Angiogenesis in Human Metastatic Malignant Melanoma. Am. J. Pathol. 2021, 191, 2023–2038. [Google Scholar] [CrossRef]
- Dome, B.; Timar, J.; Ladanyi, A.; Paku, S.; Renyi-Vamos, F.; Klepetko, W.; Lang, G.; Dome, P.; Bogos, K.; Tovari, J. Circulating Endothelial Cells, Bone Marrow-Derived Endothelial Progenitor Cells and Proangiogenic Hematopoietic Cells in Cancer: From Biology to Therapy. Crit. Rev. Oncol. Hematol. 2009, 69, 108–124. [Google Scholar] [CrossRef]
- Tasev, D.; Koolwijk, P.; Van Hinsbergh, V.W.M. Therapeutic Potential of Human-Derived Endothelial Colony-Forming Cells in Animal Models. Tissue Eng. Part B Rev. 2016, 22, 371–382. [Google Scholar] [CrossRef]
- Fujisawa, T.; Tura-Ceide, O.; Hunter, A.; Mitchell, A.; Vesey, A.; Medine, C.; Gallogly, S.; Hadoke, P.W.F.; Keith, C.; Sproul, A.; et al. Endothelial Progenitor Cells Do Not Originate From the Bone Marrow. Circulation 2019, 140, 1524–1526. [Google Scholar] [CrossRef]
- Biagioni, A.; Laurenzana, A.; Menicacci, B.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Guasti, D.; Paoli, P.; Serratì, S.; Mocali, A.; et al. UPAR-Expressing Melanoma Exosomes Promote Angiogenesis by VE-Cadherin, EGFR and UPAR Overexpression and Rise of ERK1,2 Signaling in Endothelial Cells. Cell. Mol. Life Sci. 2021, 78, 3057–3072. [Google Scholar] [CrossRef]
- Laurenzana, A.; Biagioni, A.; D’Alessio, S.; Bianchini, F.; Chillà, A.; Margheri, F.; Luciani, C.; Mazzanti, B.; Pimpinelli, N.; Torre, E.; et al. Melanoma Cell Therapy: Endothelial Progenitor Cells as Shuttle of the MMP12 UPAR-Degrading Enzyme. Oncotarget 2014, 5, 3711–3727. [Google Scholar] [CrossRef]
- Pan, J.; Yuan, K.; Shanshan, P.; Yanqin, H.; Yujuan, Z.; Hu, Y.; Yuanyuan, F.; Shi, Y.; Liu, Y.; Hongmei, W.; et al. Gene Silencing of Indoleamine 2,3-Dioxygenase Hinders Tumor Growth through Angiogenesis Inhibition. Int. J. Oncol. 2017, 50, 2136–2144. [Google Scholar] [CrossRef]
- Zhang, W.; Mao, S.; Shi, D.; Zhang, J.; Zhang, Z.; Guo, Y.; Wu, Y.; Wang, R.; Wang, L.; Huang, Y.; et al. MicroRNA-153 Decreases Tryptophan Catabolism and Inhibits Angiogenesis in Bladder Cancer by Targeting Indoleamine 2,3-Dioxygenase 1. Front. Oncol. 2019, 9, 619. [Google Scholar] [CrossRef]
- Wei, L.; Zhu, S.; Li, M.; Li, F.; Wei, F.; Liu, J.; Ren, X. High Indoleamine 2,3-Dioxygenase Is Correlated With Microvessel Density and Worse Prognosis in Breast Cancer. Front. Immunol. 2018, 9, 724. [Google Scholar] [CrossRef]
- Hoffmann, D.; Dvorakova, T.; Stroobant, V.; Bouzin, C.; Daumerie, A.; Solvay, M.; Klaessens, S.; Letellier, M.C.; Renauld, J.C.; van Baren, N.; et al. Tryptophan 2,3-Dioxygenase Expression Identified in Human Hepatocellular Carcinoma Cells and in Intratumoral Pericytes of Most Cancers. Cancer Immunol. Res. 2020, 8, 19–31. [Google Scholar] [CrossRef]
- Paccosi, S.; Cecchi, M.; Silvano, A.; Fabbri, S.; Parenti, A. Different Effects of Tryptophan 2,3-Dioxygenase Inhibition on SK-Mel-28 and HCT-8 Cancer Cell Lines. J. Cancer Res. Clin. Oncol. 2020, 146, 3155–3163. [Google Scholar] [CrossRef]
- Margheri, F.; Chillà, A.; Laurenzana, A.; Serratì, S.; Mazzanti, B.; Saccardi, R.; Santosuosso, M.; Danza, G.; Sturli, N.; Rosati, F.; et al. Endothelial Progenitor Cell-Dependent Angiogenesis Requires Localization of the Full-Length Form of UPAR in Caveolae. Blood 2011, 118, 3743–3755. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Rossi, S.; Cordella, M.; Tabolacci, C.; Nassa, G.; D’Arcangelo, D.; Senatore, C.; Pagnotto, P.; Magliozzi, R.; Salvati, A.; Weisz, A.; et al. TNF-Alpha and Metalloproteases as Key Players in Melanoma Cells Aggressiveness. J. Exp. Clin. Cancer Res. 2018, 37, 326. [Google Scholar] [CrossRef]
- Goździalska, A.; Wojas-Pelc, A.; Drąg, J.; Brzewski, P.; Jaśkiewicz, J.; Pastuszczak, M. Expression of Metalloproteinases (MMP-2 and MMP-9) in Basal-Cell Carcinoma. Mol. Biol. Rep. 2016, 43, 1027–1033. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Tumor Angiogenesis: MMP-Mediated Induction of Intravasation- and Metastasis-Sustaining Neovasculature. Matrix Biol. 2015, 44–46, 94–112. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Overcoming Immune Evasion in Melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar] [CrossRef]
- Filippi, L.; Bruno, G.; Domazetovic, V.; Favre, C.; Calvani, M. Current Therapies and New Targets to Fight Melanoma: A Promising Role for the Β3-Adrenoreceptor. Cancers 2020, 12, 1415. [Google Scholar] [CrossRef]
- Ramelyte, E.; Schindler, S.A.; Dummer, R. The Safety of Anti PD-1 Therapeutics for the Treatment of Melanoma. Expert Opin. Drug Saf. 2017, 16, 41–53. [Google Scholar] [CrossRef]
- Liu, Q.; Nie, R.; Li, M.; Li, L.; Zhou, H.; Lu, H.; Wang, X. Identification of Subtypes Correlated with Tumor Immunity and Immunotherapy in Cutaneous Melanoma. Comput. Struct. Biotechnol. J. 2021, 19, 4472–4485. [Google Scholar] [CrossRef]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation of a Tumor Factor Responsible for Angiogenesis. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-Angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lin, Y.; Wang, C.; Deng, L.; Chen, M.; Assaraf, Y.G.; Chen, Z.S.; Ye, W.; Zhang, D. New Insights into Antiangiogenic Therapy Resistance in Cancer: Mechanisms and Therapeutic Aspects. Drug Resist. Updat. 2022, 64, 100849. [Google Scholar] [CrossRef]
- Vasudev, N.S.; Reynolds, A.R. Anti-Angiogenic Therapy for Cancer: Current Progress, Unresolved Questions and Future Directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef]
- Nonaka, H.; Saga, Y.; Fujiwara, H.; Akimoto, H.; Yamada, A.; Kagawa, S.; Takei, Y.; Machida, S.; Takikawa, O.; Suzuki, M. Indoleamine 2,3-Dioxygenase Promotes Peritoneal Dissemination of Ovarian Cancer through Inhibition of Natural Killercell Function and Angiogenesis Promotion. Int. J. Oncol. 2011, 38, 113–120. [Google Scholar] [CrossRef]
- Brouillet, S.; Hoffmann, P.; Benharouga, M.; Salomon, A.; Schaal, J.P.; Feige, J.J.; Alfaidy, N. Molecular Characterization of EG-VEGF-Mediated Angiogenesis: Differential Effects on Microvascular and Macrovascular Endothelial Cells. Mol. Biol. Cell 2010, 21, 2832–2843. [Google Scholar] [CrossRef]
- Zabroski, I.O.; Nugent, M.A. Lipid Raft Association Stabilizes VEGF Receptor 2 in Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 798. [Google Scholar] [CrossRef]
- Folgiero, V.; Miele, E.; Carai, A.; Ferretti, E.; Alfano, V.; Po, A.; Bertaina, V.; Goffredo, B.M.; Benedetti, M.C.; Camassei, F.D.; et al. IDO1 Involvement in MTOR Pathway: A Molecular Mechanism of Resistance to MTOR Targeting in Medulloblastoma. Oncotarget 2016, 7, 52900–52911. [Google Scholar] [CrossRef]
- Barati, M.; Akhondi, M.; Mousavi, N.S.; Haghparast, N.; Ghodsi, A.; Baharvand, H.; Ebrahimi, M.; Hassani, S.N. Pluripotent Stem Cells: Cancer Study, Therapy, and Vaccination. Stem Cell Rev. Rep. 2021, 17, 1975–1992. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, A.G.; Tessema, K.; Muthukrishnan, S.D.; Sober, M.; Kawaguchi, R.; Laks, D.R.; Bhaduri, A.; Swarup, V.; Nathanson, D.A.; Geschwind, D.H.; et al. Pathway-Based Approach Reveals Differential Sensitivity to E2F1 Inhibition in Glioblastoma. Cancer Res. Commun. 2022, 2, 1049–1060. [Google Scholar] [CrossRef]
- Alla, V.; Engelmann, D.; Niemetz, A.; Pahnke, J.; Schmidt, A.; Kunz, M.; Emmrich, S.; Steder, M.; Koczan, D.; Pützer, B.M. E2F1 in Melanoma Progression and Metastasis. J. Natl. Cancer Inst. 2010, 102, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K. Emerging Evidence for Krüppel-Like Factor 4 (KLF4) as a Tumor Suppressor in Neuroblastoma. In Neuroblastoma; Molecular Mechanisms and Therapeutic Interventions; Academic Press: Cambridge, MA, USA, 2019; pp. 283–297. [Google Scholar] [CrossRef]
- He, Z.; He, J.; Xie, K. KLF4 Transcription Factor in Tumorigenesis. Cell Death Discov. 2023, 9, 118. [Google Scholar] [CrossRef]
- Kim, J.; Liu, Y.; Qiu, M.; Xu, Y. Pluripotency Factor Nanog Is Tumorigenic by Deregulating DNA Damage Response in Somatic Cells. Oncogene 2016, 35, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Periván, S.I.; Gómez-Gómez, Y.; Leyva-Vázquez, M.A.; Lagunas-Martínez, A.; Organista-Nava, J.; Illades-Aguiar, B. Role of Oct3/4 in Cervical Cancer Tumorigenesis. Front. Oncol. 2020, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Qin, K.; Wang, D.; Zhang, Y.; Bai, N.; Yang, S.; Luo, Y.; Xiang, R.; Tan, X. Overexpression of Oct4 Suppresses the Metastatic Potential of Breast Cancer Cells via Rnd1 Downregulation. Biochim. Biophys. Acta 2014, 1842, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Jia, Y.; Qin, T.; Zhang, C.; Li, Y.; Huang, C.; Liu, Z.; Wang, J.; Li, K. Bevacizumab Promotes Active Biological Behaviors of Human Umbilical Vein Endothelial Cells by Activating TGFβ1 Pathways via Off-VEGF Signaling. Cancer Biol. Med. 2020, 17, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Corselli, M.; Parodi, A.; Mogni, M.; Sessarego, N.; Kunkl, A.; Dagna-Bricarelli, F.; Ibatici, A.; Pozzi, S.; Bacigalupo, A.; Frassoni, F.; et al. Clinical Scale Ex Vivo Expansion of Cord Blood-Derived Outgrowth Endothelial Progenitor Cells Is Associated with High Incidence of Karyotype Aberrations. Exp. Hematol. 2008, 36, 340–349. [Google Scholar] [CrossRef]
- Laurenzana, A.; Margheri, F.; Biagioni, A.; Chillà, A.; Pimpinelli, N.; Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Calorini, L.; Serratì, S.; et al. EGFR/UPAR Interaction as Druggable Target to Overcome Vemurafenib Acquired Resistance in Melanoma Cells. eBioMedicine 2019, 39, 194–206. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cecchi, M.; Anceschi, C.; Silvano, A.; Coniglio, M.L.; Chinnici, A.; Magnelli, L.; Lapucci, A.; Laurenzana, A.; Parenti, A. Unveiling the Role of Tryptophan 2,3-Dioxygenase in the Angiogenic Process. Pharmaceuticals 2024, 17, 558. https://0-doi-org.brum.beds.ac.uk/10.3390/ph17050558
Cecchi M, Anceschi C, Silvano A, Coniglio ML, Chinnici A, Magnelli L, Lapucci A, Laurenzana A, Parenti A. Unveiling the Role of Tryptophan 2,3-Dioxygenase in the Angiogenic Process. Pharmaceuticals. 2024; 17(5):558. https://0-doi-org.brum.beds.ac.uk/10.3390/ph17050558
Chicago/Turabian StyleCecchi, Marta, Cecilia Anceschi, Angela Silvano, Maria Luisa Coniglio, Aurora Chinnici, Lucia Magnelli, Andrea Lapucci, Anna Laurenzana, and Astrid Parenti. 2024. "Unveiling the Role of Tryptophan 2,3-Dioxygenase in the Angiogenic Process" Pharmaceuticals 17, no. 5: 558. https://0-doi-org.brum.beds.ac.uk/10.3390/ph17050558