
Mitochondria-Derived Vesicles, Sterile Inflammation, and Pyroptosis in Liver Cancer: Partners in Crime or Innocent Bystanders?
 , , and
, , and 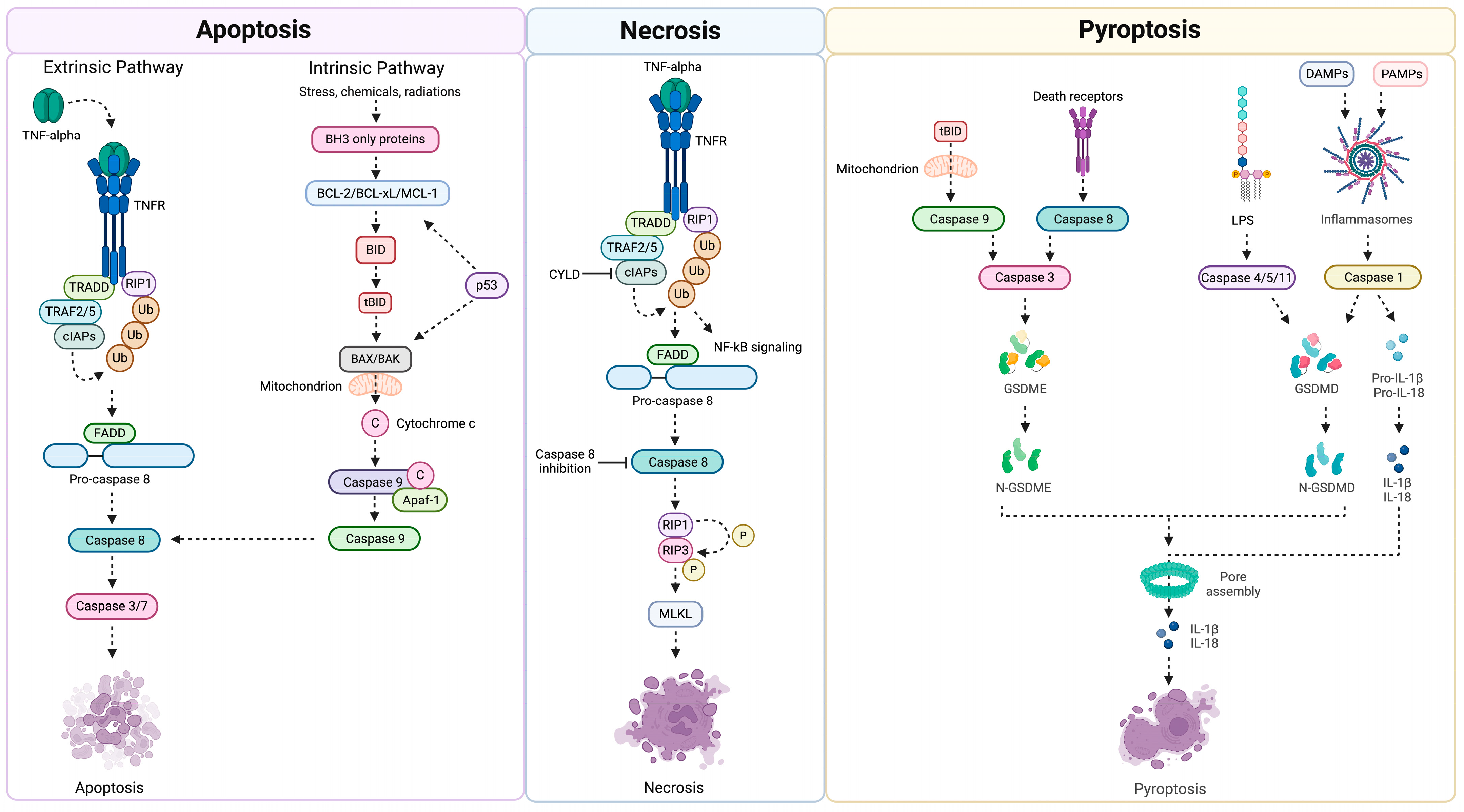{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Mechanisms Linking Mitochondrial Dysfunction and Inflammation
2.1. Mechanisms of Cell Death: Apoptosis Versus Necrosis
2.2. Pro-Inflammatory Apoptosis: Pyroptosis and Its Role in the Pathogenesis of Hepatocarcinoma
3. Mitochondria-Derived Vesicles in Hepatocarcinoma: Knowing the Unknown
4. From Bench to Bedside: Clinical Management of Hepatocarcinoma and Immunotherapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory mechanisms of HCC development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Huang, S.; Berger, E.; Liu, L.; Gross, N.; Heinzmann, F.; Ringelhan, M.; Connor, T.O.; Stadler, M.; Meister, M.; et al. Kupffer cell-derived Tnf triggers cholangiocellular tumorigenesis through JNK Due to chronic mitochondrial dysfunction and ROS. Cancer Cell 2017, 31, 771–789.e6. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Romano, R.; Bucci, C.; Marzetti, E. Extracellular vesicles and damage-associated molecular patterns: A Pandora’s box in health and disease. Front. Immunol. 2020, 11, 601740. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Primiano, A.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; et al. Circulating mitochondrial-derived vesicles, inflammatory biomarkers and amino acids in older adults with physical frailty and sarcopenia: A preliminary BIOSPHERE multi-marker study using sequential and orthogonalized covariance selection—Linear discriminant analysis. Front. Cell Dev. Biol. 2020, 8, 564417. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.; Bucci, C.; Marzetti, E. Circulating Extracellular Vesicles: Friends and Foes in Neurodegeneration. Neural Regen. Res. 2022, 17, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Zhang, L.; Wu, M.; Cao, K.; Jiang, F.; Chen, D.; Li, N.; Li, W. The significance of exosomes in the development and treatment of hepatocellular carcinoma. Mol. Cancer 2020, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Pucci, M.; Alessandro, R.; Fontana, S. Extracellular vesicles and tumor-immune escape: Biological functions and clinical perspectives. Int. J. Mol. Sci. 2020, 21, 2286. [Google Scholar] [CrossRef]
- Deng, M.; Sun, S.; Zhao, R.; Guan, R.; Zhang, Z.; Li, S.; Wei, W.; Guo, R. The pyroptosis-related gene signature predicts prognosis and indicates immune activity in hepatocellular carcinoma. Mol. Med. 2022, 28, 16. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, L.-C.; Guan, Y.; Ho, M.-C.; Lu, S.; Spahn, J.; Hsu, C.-H. Atezolizumab plus bevacizumab combination enables an unresectable hepatocellular carcinoma resectable and links immune exclusion and tumor dedifferentiation to acquired resistance. Exp. Hematol. Oncol. 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Scheiner, B.; Peck-Radosavljevic, M. Immunotherapy for advanced hepatocellular carcinoma: A focus on special subgroups. Gut 2021, 70, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, R.; Kheirollahi, A.; Davoodi, J. Apaf-1: Regulation and function in cell death. Biochimie 2017, 135, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.K.; Wang, L.; Zhang, P. Antitumor effects of Chrysanthemin in PC-3 human prostate cancer cells are mediated via apoptosis induction, caspase signalling pathway and loss of mitochondrial membrane potential. Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.F.; Abd El-Hafeez, A.A.; Abbas, S.H.; Abdelhamid, D.; Abdel-Aziz, M. New 1,2,4-triazole-chalcone hybrids induce caspase-3 dependent apoptosis in A549 human lung adenocarcinoma cells. Eur. J. Med. Chem. 2018, 151, 705–722. [Google Scholar] [CrossRef] [PubMed]
- Kayacan, S.; Sener, L.T.; Melikoglu, G.; Kultur, S.; Albeniz, I.; Ozturk, M. Induction of apoptosis by Centaurea nerimaniae extract in HeLa and MDA-MB-231 cells by a caspase-3 pathway. Biotech. Histochem. 2018, 93, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-harbi, S.; Almasan, A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. In Apoptosis and Cancer; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1219, pp. 1–9. [Google Scholar] [CrossRef]
- Shaham, S.; Horvitz, H.R. Developing Caenorhabditis elegans neurons may contain both cell-death protective and killer activities. Genes Dev. 1996, 10, 578–591. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Syntichaki, P.; Xu, K.; Driscoll, M.; Tavernarakis, N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 2002, 419, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell. 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Kitsis, R.N.; Molkentin, J.D. Apoptotic cell death “Nixed” by an ER-mitochondrial necrotic pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 9031–9032. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.Y.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2013, e00772. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Talà, A.; Guerra, F.; Calcagnile, M.; Romano, R.; Resta, S.C.; Paiano, A.; Chiariello, M.; Pizzolante, G.; Bucci, C.; Alifano, P. HrpA anchors meningococci to the dynein motor and affects the balance between apoptosis and pyroptosis. J. Biomed. Sci. 2022, 29, 45. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.-X.; Dai, Y.-Z.; Zhao, Y.-Z.; Nie, K. Gasdermin E: A prospective target for therapy of diseases. Front. Pharmacol. 2022, 13, 855828. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, Y.; Tan, X.; Merkher, Y.; Leonov, S.; Zhu, L.; Deng, Y.; Zhang, H.; Zhu, D.; Tan, Y.; et al. Emerging mechanisms of pyroptosis and its therapeutic strategy in cancer. Cell Death Discov. 2022, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- de Torre-Minguela, C.; Gómez, A.I.; Couillin, I.; Pelegrín, P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J. 2021, 35, e21757. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef]
- Wang, J.L.; Hua, S.N.; Bao, H.J.; Yuan, J.; Zhao, Y.; Chen, S. Pyroptosis and inflammasomes in cancer and inflammation. MedComm 2023, 4, e374. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Ou, Z.; Zhang, W.; Zhu, X.; Li, P.; Gong, J. Cathepsin B regulates non-canonical NLRP3 inflammasome pathway by modulating activation of caspase-11 in Kupffer cells. Cell Prolif. 2018, 51, e12487. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kanneganti, T.-D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Faitg, J.; Auwerx, J.; Ferrucci, L.; D’Amico, D. Mitophagy in human health, ageing and disease. Nat. Metab. 2023, 5, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; Alaarg, A.; Smith, C.I.E.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed]
- Royo, F.; Théry, C.; Falcón-Pérez, J.M.; Nieuwland, R.; Witwer, K.W. Methods for separation and characterization of extracellular vesicles: Results of a worldwide survey performed by the ISEV rigor and standardization subcommittee. Cells 2020, 9, 1955. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and ectosomes in intercellular communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bucci, C.; Marzetti, E. Mitochondrial-derived vesicles: The good, the bad, and the ugly. Int. J. Mol. Sci. 2023, 24, 13835. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Junior, H.J.; Damiano, F.P.; Bucci, C.; Marzetti, E. Circulating mitochondrial DNA and inter-organelle contact sites in aging and associated conditions. Cells 2022, 11, 675. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Beli, R.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Bucci, C.; Guerra, F.; Marzetti, E. Older adults with physical frailty and sarcopenia show increased levels of circulating small extracellular vesicles with a specific mitochondrial signature. Cells 2020, 9, 973. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.R.; et al. Mitochondrial signatures in circulating extracellular vesicles of older adults with Parkinson’s disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and release of mitochondrial-derived vesicles in health, aging and disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Cadete, V.J.J.; Deschênes, S.; Cuillerier, A.; Brisebois, F.; Sugiura, A.; Vincent, A.; Turnbull, D.; Picard, M.; McBride, H.M.; Burelle, Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 2016, 594, 5343–5362. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 Function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Vasam, G.; Nadeau, R.; Cadete, V.J.J.; Lavallée-Adam, M.; Menzies, K.J.; Burelle, Y. Proteomics characterization of mitochondrial-derived vesicles under oxidative stress. FASEB J. 2021, 35, e21278. [Google Scholar] [CrossRef] [PubMed]
- Abuaita, B.H.; Schultz, T.L.; O’Riordan, M.X. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, H.; Wu, Y.; Zhu, Y.; Zhang, J.; Yang, G.; Yan, Q.; Li, J.; Li, T.; Liu, L. Mitochondrial-derived vesicles protect cardiomyocytes against hypoxic damage. Front. Cell Dev. Biol. 2020, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Tang, B.L. Pyruvate dehydrogenase complex (PDC) export from the mitochondrial matrix. Mol. Membr. Biol. 2014, 31, 207–210. [Google Scholar] [CrossRef]
- Towers, C.G.; Wodetzki, D.K.; Thorburn, J.; Smith, K.R.; Caino, M.C.; Thorburn, A. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell 2021, 56, 2029–2042.e5. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; White, E. MDVs to the rescue: How autophagy-deficient cancer cells adapt to defective mitophagy. Dev. Cell 2021, 56, 2010–2012. [Google Scholar] [CrossRef]
- Mondal, P.; Towers, C. Beyond mitophagy: Mitochondrial-derived vesicles can get the job done! Autophagy 2022, 18, 449–451. [Google Scholar] [CrossRef]
- Andrade-Navarro, M.A.; Sanchez-Pulido, L.; McBride, H.M. Mitochondrial vesicles: An ancient process providing new links to peroxisomes. Curr. Opin. Cell Biol. 2009, 21, 560–567. [Google Scholar] [CrossRef]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Song, K.-X.; Qu, S. Nonalcoholic fatty liver disease: Pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Zhang, H.-X.; Guo, J.-R.; Lam, C.W.K.; Wang, C.-Y.; Zhang, W. Mitochondria-mediated pathogenesis and therapeutics for non-alcoholic fatty liver disease. Mol. Nutr. Food Res. 2019, 63, e1900043. [Google Scholar] [CrossRef]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 2021, 142, 112041. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465. [Google Scholar] [CrossRef]
- Sookoian, S.; Flichman, D.; Scian, R.; Rohr, C.; Dopazo, H.; Gianotti, T.F.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J. Pathol. 2016, 240, 437–449. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, I.; Rodríguez-Juan, C.; Díaz-Sanjuan, T.; del Hoyo, P.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591. [Google Scholar] [CrossRef]
- Liu, C.; Wu, H.; Mao, Y.; Chen, W.; Chen, S. Exosomal microRNAs in hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 254. [Google Scholar] [CrossRef]
- Wang, H.; Hou, L.; Li, A.; Duan, Y.; Gao, H.; Song, X. Expression of serum exosomal microRNA-21 in human hepatocellular carcinoma. BioMed Res. Int. 2014, 2014, 864894. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Sun, W.; Yue, S.; Yang, J.; Li, J.; Ma, B.; Wang, J.; Yang, X.; Pu, M.; et al. Loss of exosomal MiR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017, 397, 33–42. [Google Scholar] [CrossRef]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal MiR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef]
- Liu, G.; Ouyang, X.; Sun, Y.; Xiao, Y.; You, B.; Gao, Y.; Yeh, S.; Li, Y.; Chang, C. The MiR-92a-2-5p in exosomes from macrophages increases liver cancer cells invasion via altering the AR/PHLPP/p-AKT/β-catenin Signaling. Cell Death Differ. 2020, 27, 3258–3272. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, J.; Ou, S.; Chen, J.; Chen, L. Adipose-derived exosomes deliver MiR-23a/b to regulate tumor growth in hepatocellular cancer by targeting the VHL/HIF axis. J. Physiol. Biochem. 2019, 75, 391–401. [Google Scholar] [CrossRef]
- Tian, X.-P.; Wang, C.-Y.; Jin, X.-H.; Li, M.; Wang, F.-W.; Huang, W.-J.; Yun, J.-P.; Xu, R.-H.; Cai, Q.-Q.; Xie, D. Acidic microenvironment up-regulates exosomal MiR-21 and MiR-10b in early-stage hepatocellular carcinoma to promote cancer cell proliferation and metastasis. Theranostics 2019, 9, 1965–1979. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Zhang, X.; Shao, T.; Luo, Y.; Wang, W.; Han, Y. Extracellular vesicles and hepatocellular carcinoma: Opportunities and challenges. Front. Oncol. 2022, 12, 884369. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Cerreto, M.; Cardone, F.; Cerrito, L.; Stella, L.; Santopaolo, F.; Pallozzi, M.; Gasbarrini, A.; Ponziani, F.R. The New Era of Systemic Treatment for Hepatocellular Carcinoma: From the First Line to the Optimal Sequence. Curr. Oncol. 2023, 30, 8774–8792. [Google Scholar] [CrossRef] [PubMed]
- Vitale, A.; Cabibbo, G.; Iavarone, M.; Viganò, L.; Pinato, D.J.; Ponziani, F.R.; Lai, Q.; Casadei-Gardini, A.; Celsa, C.; Galati, G.; et al. Personalised management of patients with hepatocellular carcinoma: A multiparametric therapeutic hierarchy concept. Lancet Oncol. 2023, 24, e312–e322. [Google Scholar] [CrossRef] [PubMed]
- Trevisani, F.; Vitale, A.; Kudo, M.; Kulik, L.; Park, J.-W.; Pinato, D.J.; Cillo, U. Merits and boundaries of the BCLC staging and treatment algorithm: Learning from the past to improve the future with a novel proposal. J. Hepatol. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Huber, L.; Stewart, J.; Mathews, M.; Falcon, B.; Chintharlapalli, S. Evaluation of AFP expression as a predictive marker for response to anti-VEGFR-2 inhibition. Ann. Oncol. 2017, 28, iii19–iii20. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40 (Suppl. S4), 379. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.H.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Updated efficacy and safety of KEYNOTE-224: A phase ii study of pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. Eur. J. Cancer 2022, 167, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The CheckMate 040 randomized clinical trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Minici, R.; Siciliano, M.A.; Ammendola, M.; Santoro, R.C.; Barbieri, V.; Ranieri, G.; Laganà, D. Prognostic role of neutrophil-to-lymphocyte ratio (NLR), lymphocyte-to-monocyte ratio (LMR), platelet-to-lymphocyte ratio (PLR) and lymphocyte-to-C reactive protein ratio (LCR) in patients with hepatocellular carcinoma (HCC) undergoing chemoembolizations (TACE) of the liver: The unexplored corner linking tumor microenvironment, biomarkers and interventional radiology. Cancers 2022, 15, 257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Huang, H.; Shi, J.; Zhao, Y.; Dong, Q.; Jia, H.; Liu, Y.; Ye, Q.; Sun, H.; Zhu, X.; et al. Prognostic value of interleukin 2 and interleukin 15 in peritumoral hepatic tissues for patients with hepatitis B-related hepatocellular carcinoma after curative resection. Gut 2010, 59, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.H.; Jia, H.L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.X.; Tang, Z.Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef]
- Sanghera, C.; Teh, J.J.; Pinato, D.J. The systemic inflammatory response as a source of biomarkers and therapeutic targets in hepatocellular carcinoma. Liver Int. 2019, 39, 2008–2023. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, F.; Ponziani, F.R.; Cardone, F.; Bucci, C.; Marzetti, E.; Picca, A. Mitochondria-Derived Vesicles, Sterile Inflammation, and Pyroptosis in Liver Cancer: Partners in Crime or Innocent Bystanders? Int. J. Mol. Sci. 2024, 25, 4783. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094783
Guerra F, Ponziani FR, Cardone F, Bucci C, Marzetti E, Picca A. Mitochondria-Derived Vesicles, Sterile Inflammation, and Pyroptosis in Liver Cancer: Partners in Crime or Innocent Bystanders? International Journal of Molecular Sciences. 2024; 25(9):4783. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094783
Chicago/Turabian StyleGuerra, Flora, Francesca Romana Ponziani, Ferdinando Cardone, Cecilia Bucci, Emanuele Marzetti, and Anna Picca. 2024. "Mitochondria-Derived Vesicles, Sterile Inflammation, and Pyroptosis in Liver Cancer: Partners in Crime or Innocent Bystanders?" International Journal of Molecular Sciences 25, no. 9: 4783. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094783