
Gene Expression and DNA Methylation Profiling Suggest Potential Biomarkers for Azacitidine Resistance in Myelodysplastic Syndrome


Abstract
:1. Introduction
2. Results
2.1. Levels of DNMTs and Methyl-Binding Proteins Are Higher in F-36P/AZA Cells than in F-36P Cells
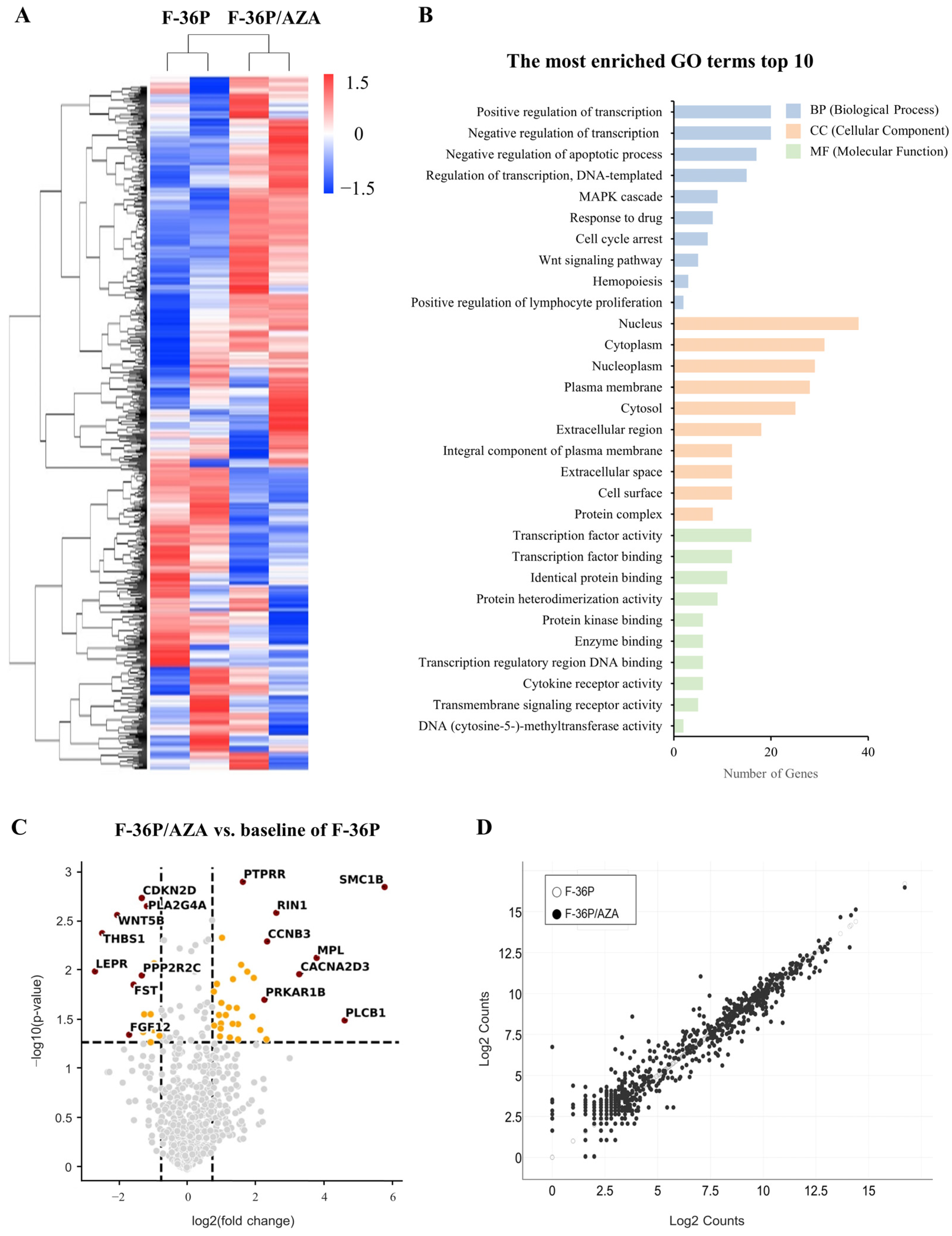
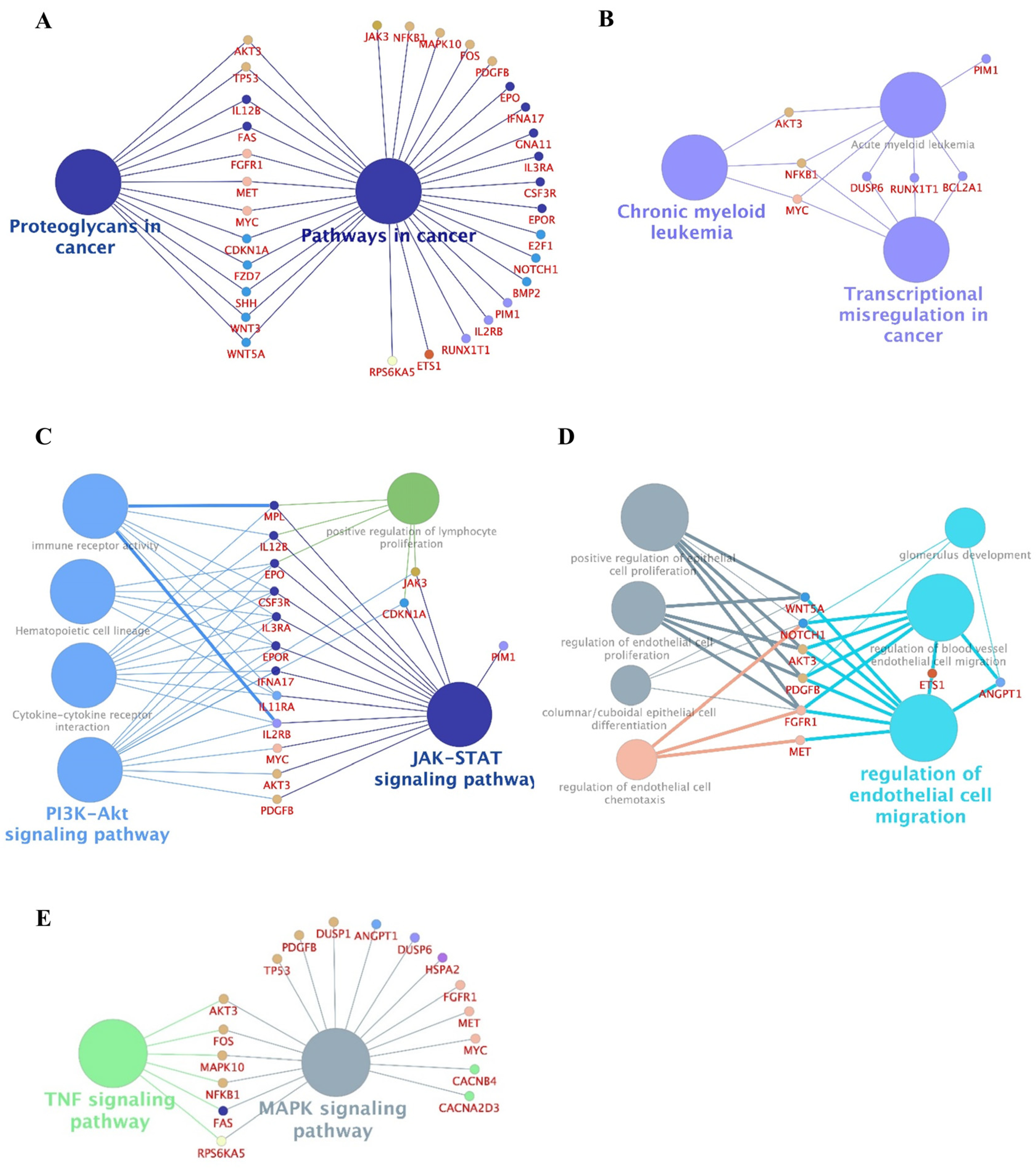
2.2. Identification of Genes Exhibiting Differential Expression between F-36P/AZA and F-36P Cells
2.3. Evaluation of Candidate Genes via qRT-PCR and Pyrosequencing
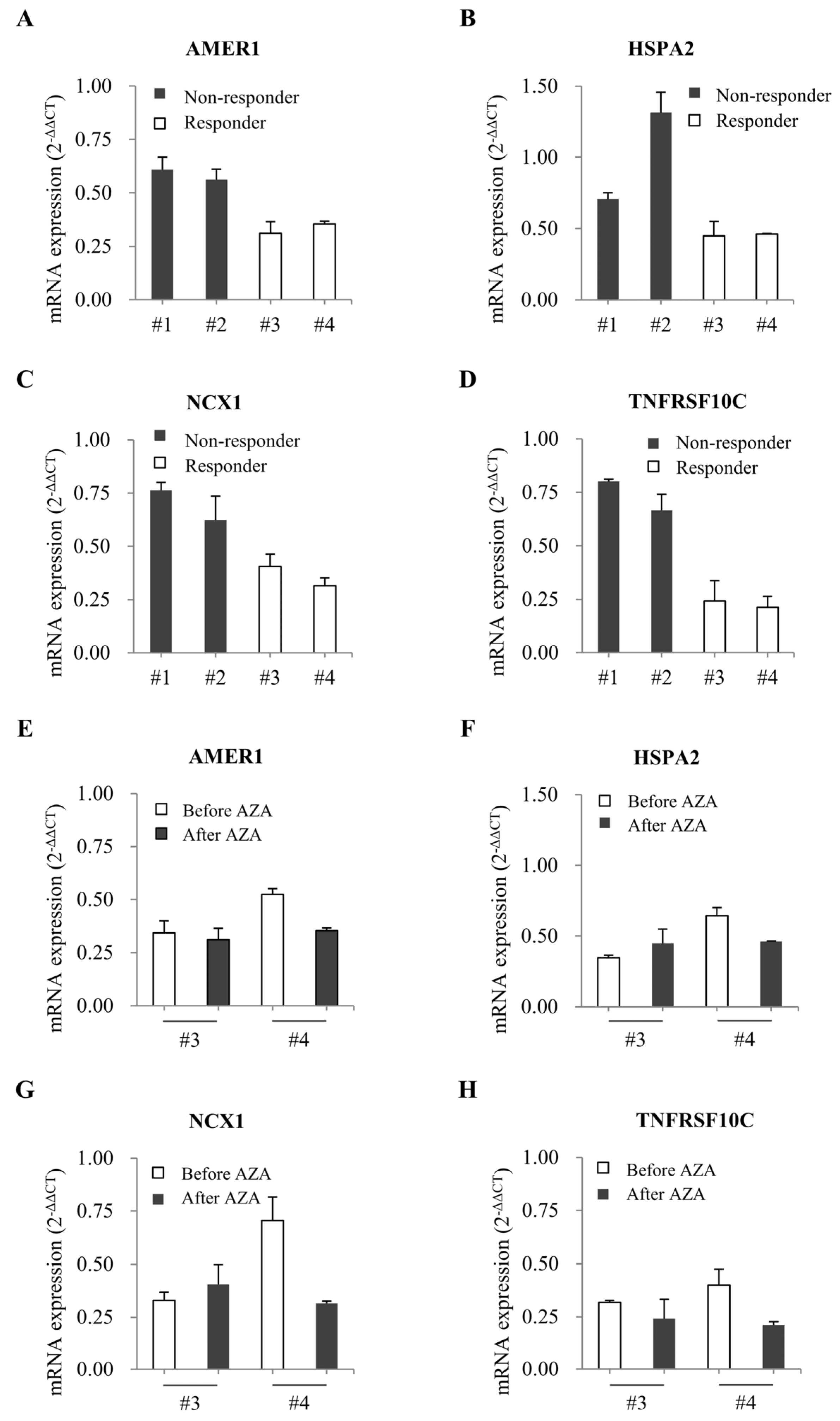
2.4. Comparison of Gene Expression in Bone Marrow from Patients with MDS
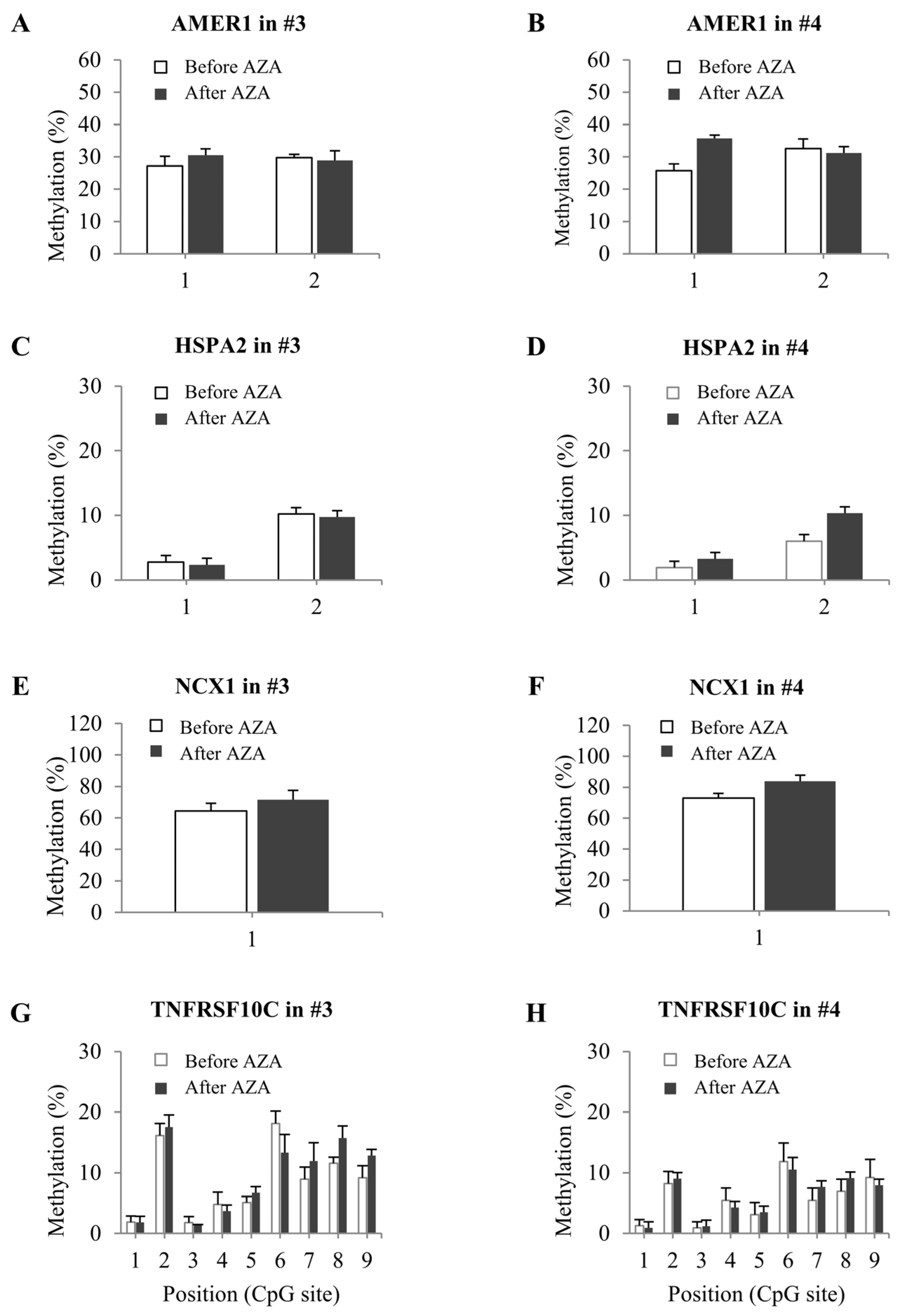
2.5. DNA Methylation Status in Bone Marrow from Patients with MDS
3. Discussion
4. Materials and Methods
4.1. AZA-Resistant Cell Selection and Culture
4.2. RNA Isolation
4.3. Quantitative Reverse Transcription-PCR (qRT-PCR)
4.4. Western Blotting
4.5. NanoString Gene Expression Analysis
4.6. Gene Ontology (GO) and Pathway Enrichment Analyses of Differentially Expressed Genes (DEGs)
4.7. Protein–Protein Network and Module Analyses
4.8. DNA Extraction and Bisulfite Conversion
4.9. Quantitative Methylation Analysis via Pyrosequencing
4.10. Validation of Genetic Alterations in Candidate Genes
4.11. Patient Enrollment and Treatment
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cazzola, M. Myelodysplastic syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Le, Y. Screening and identification of key candidate genes and pathways in myelodysplastic syndrome by bioinformatic analysis. PeerJ 2019, 7, e8162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yin, H.; Li, N.; Zhu, Y.; Shen, W.; Qian, S.; He, G.; Li, J.; Wang, X. An integrated regulatory network based on comprehensive analysis of mRNA expression, gene methylation and expression of long non-coding RNAs (lncRNAs) in myelodysplastic syndromes. Front. Oncol. 2019, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Ball, B.; Zeidan, A.; Gore, S.D.; Prebet, T. Hypomethylating agent combination strategies in myelodysplastic syndromes: Hopes and shortcomings. Leuk. Lymphoma 2017, 58, 1022–1036. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Haase, D.; Santini, V.; Sanz, G.F.; Platzbecker, U.; Mey, U. Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Belasen, A.; Navada, S. Therapeutic Options in Myelodysplastic Syndromes Following Hypomethylating Agent Failure. J. Sci. Innov. Med. 2020, 3, 52–64. [Google Scholar] [CrossRef]
- Krejcik, Z.; Belickova, M.; Hrustincova, A.; Votavova, H.; Jonasova, A.; Cermak, J.; Dyr, J.E.; Merkerova, M.D. MicroRNA profiles as predictive markers of response to azacitidine therapy in myelodysplastic syndromes and acute myeloid leukemia. Cancer Biomark. 2018, 22, 101–110. [Google Scholar] [CrossRef]
- Nazha, A.; Sekeres, M.A.; Bejar, R.; Rauh, M.J.; Othus, M.; Komrokji, R.S.; Barnard, J.; Hilton, C.B.; Kerr, C.M.; Steensma, D.P. Genomic biomarkers to predict resistance to hypomethylating agents in patients with myelodysplastic syndromes using artificial intelligence. JCO Precis. Oncol. 2019, 3, PO.19.00119. [Google Scholar] [CrossRef] [PubMed]
- Meldi, K.; Qin, T.; Buchi, F.; Droin, N.; Sotzen, J.; Micol, J.-B.; Selimoglu-Buet, D.; Masala, E.; Allione, B.; Gioia, D. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J. Clin. Investig. 2015, 125, 1857–1872. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Garcia-Manero, G.; Jabbour, E. Therapeutic choices after hypomethylating agent resistance for myelodysplastic syndromes. Curr. Opin. Hematol. 2018, 25, 146–153. [Google Scholar] [CrossRef]
- Lee, E.-J.; Zeidan, A.M. Genome sequencing in myelodysplastic syndromes: Can molecular mutations predict benefit from hypomethylating agent therapy? Expert Rev. Hematol. 2015, 8, 155–158. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Lv, N.; Li, Y.; Wang, L.; Yu, L. Predictors of clinical responses to hypomethylating agents in acute myeloid leukemia or myelodysplastic syndromes. Ann. Hematol. 2018, 97, 2025–2038. [Google Scholar] [CrossRef]
- Cabezón, M.; Malinverni, R.; Bargay, J.; Xicoy, B.; Marcé, S.; Garrido, A.; CETLAM Group. Different methylation signatures at diagnosis in patients with high-risk myelodysplastic syndromes and secondary acute myeloid leukemia predict azacitidine response and longer survival. Clin. Epigenetics 2021, 13, 1–14. [Google Scholar] [CrossRef]
- Gill, H. Adding the epigenomic signature to the prognostic jigsaw of myelodysplastic neoplasm? Br. J. Haematol. 2024, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.R.; Lee, H.J.; Enjeti, A.K. Unravelling the epigenome of myelodysplastic syndrome: Diagnosis, prognosis, and response to therapy. Cancers 2020, 12, 3128. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, G.M.; Alves, J.; Reckziegel, L.; Lazzaretti, G.; Abujamra, A.L. DNA methylation events as markers for diagnosis and management of acute myeloid leukemia and myelodysplastic syndrome. Dis. Markers 2017, 2017, 5472893. [Google Scholar] [CrossRef]
- Lamim Lovatel, V.; de Souza Fernandez, C.; Ferreira Rodrigues, E.; de Cassia Tavares, R.; Sobral da Costa, E.; Abdelhay, E.; Coelho Soares Lima, S.; de Souza Fernandez, T. Expression profiles of DNA methylation and demethylation machinery components in pediatric myelodysplastic syndrome: Clinical implications. Cancer Manag. Res. 2020, 12, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Giri, A.K.; Aittokallio, T. DNMT inhibitors increase methylation in the cancer genome. Front. Pharmacol. 2019, 10, 385. [Google Scholar] [CrossRef]
- Kulkarni, M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol. 2011, 94, 25B-10. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Consortium, G.O. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Fontanillo, C. Protein–protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [PubMed]
- Osterman, T.J.; Terry, M.; Miller, R.S. Improving cancer data interoperability: The promise of the Minimal Common Oncology Data Elements (mCODE) initiative. JCO Clin. Cancer Inform. 2020, 4, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Pivkova-Veljanovska, A.; Panovska-Stavridis, I.; Chadievski, L.; Trajkova, S.; Popova-Labachevska, M.; Mojsovska, T.; Stojanoska, S.; Ridova, N.; Krstevska-Balkanov, S.; Stojanoski, Z. Haploidentical Stem Cell Transplantation in Patients with Myelodysplastic Syndrome: Case Report First Experience. Open Access Maced. J. Med. Sci. (OAMJMS) 2021, 9, 250–253. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Karimi Kelaye, S.; Najafi, F.; Kazemi, B.; Foruzandeh, Z.; Seif, F.; Solali, S.; Alivand, M.-R. The contributing factors of resistance or sensitivity to epigenetic drugs in the treatment of AML. Clin. Transl. Oncol. 2022, 24, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.-B.; Liu, Z.-T.; Wang, H.-Z.; Guo, X.-Q.; Xu, Y.-G.; Hu, X.-M. Arsenic disulfide promoted hypomethylation by increasing DNA methyltransferases expression in myelodysplastic syndrome. Drug Des. Dev. Ther. 2020, 1641–1650. [Google Scholar] [CrossRef]
- Wong, K.K.; Lawrie, C.H.; Green, T.M. Oncogenic roles and inhibitors of DNMT1, DNMT3A, and DNMT3B in acute myeloid leukaemia. Biomark. Insights 2019, 14, 1177271919846454. [Google Scholar] [CrossRef]
- Cluzeau, T.; Dubois, A.; Jacquel, A.; Luciano, F.; Renneville, A.; Preudhomme, C.; Karsenti, J.M.; Mounier, N.; Rohrlich, P.; Raynaud, S. Phenotypic and genotypic characterization of azacitidine-sensitive and resistant SKM1 myeloid cell lines. Oncotarget 2014, 5, 4384. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.K.; Nguyen, A.; Shi, T.; Tang, L.; Ni, X.; Escoubet, L.; MacBeth, K.J.; DiMartino, J.; Wells, J.A. Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc. Natl. Acad. Sci. USA 2019, 116, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Solly, F.; Koering, C.; Mohamed, A.M.; Maucort-Boulch, D.; Robert, G.; Auberger, P.; Flandrin-Gresta, P.; Ades, L.; Fenaux, P.; Kosmider, O. An miRNA–DNMT1 Axis Is Involved in Azacitidine Resistance and Predicts Survival in Higher-Risk Myelodysplastic Syndrome and Low Blast Count Acute Myeloid Leukemia. Clin. Cancer Res. 2017, 23, 3025–3034. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Fabiani, E.; Kwon, J.; Gao, C.; Falconi, G.; Valentini, L.; Gurnari, C.; Liu, Y.V.; Jones, A.I.; Yang, J. Demethylation and upregulation of an oncogene post hypomethylating treatment. MedRxiv 2020. [Google Scholar] [CrossRef]
- Lin, J.; Qian, J.; Yao, D.-m.; Qian, W.; Yang, J.; Wang, C.-z.; Chai, H.-y.; Ma, J.-c.; Deng, Z.-q.; Li, Y. Aberrant hypomethylation of SALL4 gene in patients with myelodysplastic syndrome. Leuk. Res. 2013, 37, 71–75. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H.; Carlos-Reyes, A. Role of DNA methylation in the resistance to therapy in solid tumors. Front. Oncol. 2020, 10, 1152. [Google Scholar] [CrossRef] [PubMed]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- Byun, J.M.; Yoo, S.-J.; Kim, H.-J.; Ahn, J.-S.; Koh, Y.; Jang, J.H.; Yoon, S.-S. IDH1/2 mutations in acute myeloid leukemia. Blood Res. 2022, 57, 13–19. [Google Scholar] [CrossRef]
- Ahn, J.-S.; Kim, H.-J. FLT3 mutations in acute myeloid leukemia: A review focusing on clinically applicable drugs. Blood Res. 2022, 57, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Asano, M.; Ohyashiki, J.H.; Kobayashi-Kawana, C.; Umezu, T.; Imanishi, S.; Azuma, K.; Akahane, D.; Fujimoto, H.; Ito, Y.; Ohyashiki, K. A novel non-invasive monitoring assay of 5-azacitidine efficacy using global DNA methylation of peripheral blood in myelodysplastic syndrome. Drug Des. Dev. Ther. 2019, 13, 1821–1833. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- Issa, J.-P.J.; Kantarjian, H.M.; Kirkpatrick, P. Azacitidine. Nat. Rev. Drug Discov. 2005, 4, 275. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.U. The role of next-generation sequencing in hematologic malignancies. Blood Res. 2024, 59, 11. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Takaku, F.; Tange, T.; Shibuya, K.; Misawa, C.; Sasaki, K.; Miyagawa, K.; Yazaki, Y.; Hirai, H. Establishment and erythroid differentiation of a cytokine-dependent human leukemic cell line F-36: A parental line requiring granulocyte-macrophage colony-stimulating factor or interleukin-3, and a subline requiring erythropoietin. Blood 1991, 78, 2261–2268. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K. The 5th edition of the World Health Organization classification of haematolymphoid tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Sex | Female | Male | Female | Female |
| Age (years) | 54 | 78 | 65 | 47 |
| Weight (kg) | 53.2 | 67 | 66 | 64.5 |
| Height (m) | 1.54 | 1.75 | 1.57 | 1.62 |
| BMI (Kg/m2) | 22.4 | 21.9 | 26.8 | 24.6 |
| Underlying disease | Atrial septal defect | HTN | HTN/depression | ADPKD, CKD |
| Baseline clinical characteristics | ||||
| WBC (×106/L) | 4500 | 3730 | 940 | 3470 |
| ANC (×106/L) | 1260 | 1828 | 190 | 2110 |
| Hb (g/dL) | 9.5 | 8 | 9.5 | 7.4 |
| Platelets (×109/L) | 249 | 178 | 31 | 104 |
| BM blasts (%) | 0.5 | 13.9 | 6.5 | 0.6 |
| Cytogenetic abnormalities | none | none | none | none |
| IPSS | 0 | 1.5 | 1 | 0 |
| IPSS risk category | Low | Int-1 | Int-1 | Low |
| IPSS-R | 2 | 5 | 5.5 | 2.5 |
| IPSS-R risk category | Low | High | High | Low |
| MDS subtypes (WHO) | MDS-SLD | MDS-EB2 | MDS-EB1 | MDS-SLD |
| Treatment cycle of AZA | 4 | 7 | 18 | 46 |
| Best response | NR | CR | CR | HI |
| Progression (Leukemic transformation) | No | Yes | Yes | No |
| PFS (month) | 7Y | 6M | 3Y10M | 6Y2M |
| Allogeneic HSCT | Yes | No | No | No |
| Time to HSCT | 6Y4M | |||
| F/U period (month) | 7Y | 1Y | 3Y9M | 6Y2M |
| F/U result | Dead | Dead | Dead | continued response to AZA |
| Cause of death | Pneumonia | AML | AML | Alive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.Y.; Shin, D.-Y.; Oh, S.; Kim, I.; Kim, E.J. Gene Expression and DNA Methylation Profiling Suggest Potential Biomarkers for Azacitidine Resistance in Myelodysplastic Syndrome. Int. J. Mol. Sci. 2024, 25, 4723. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094723
Kim DY, Shin D-Y, Oh S, Kim I, Kim EJ. Gene Expression and DNA Methylation Profiling Suggest Potential Biomarkers for Azacitidine Resistance in Myelodysplastic Syndrome. International Journal of Molecular Sciences. 2024; 25(9):4723. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094723
Chicago/Turabian StyleKim, Da Yeon, Dong-Yeop Shin, Somi Oh, Inho Kim, and Eun Ju Kim. 2024. "Gene Expression and DNA Methylation Profiling Suggest Potential Biomarkers for Azacitidine Resistance in Myelodysplastic Syndrome" International Journal of Molecular Sciences 25, no. 9: 4723. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094723