

Development of TRIB3-Based Therapy as a Gene-Independent Approach to Treat Retinal Degenerative Disorders
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Targeting TRIB3-AKT in Mice Alleviates Retinal Degeneration
2.2. Treatment of 661W Cells with Afatinib Reduces EGFR and TRIB3
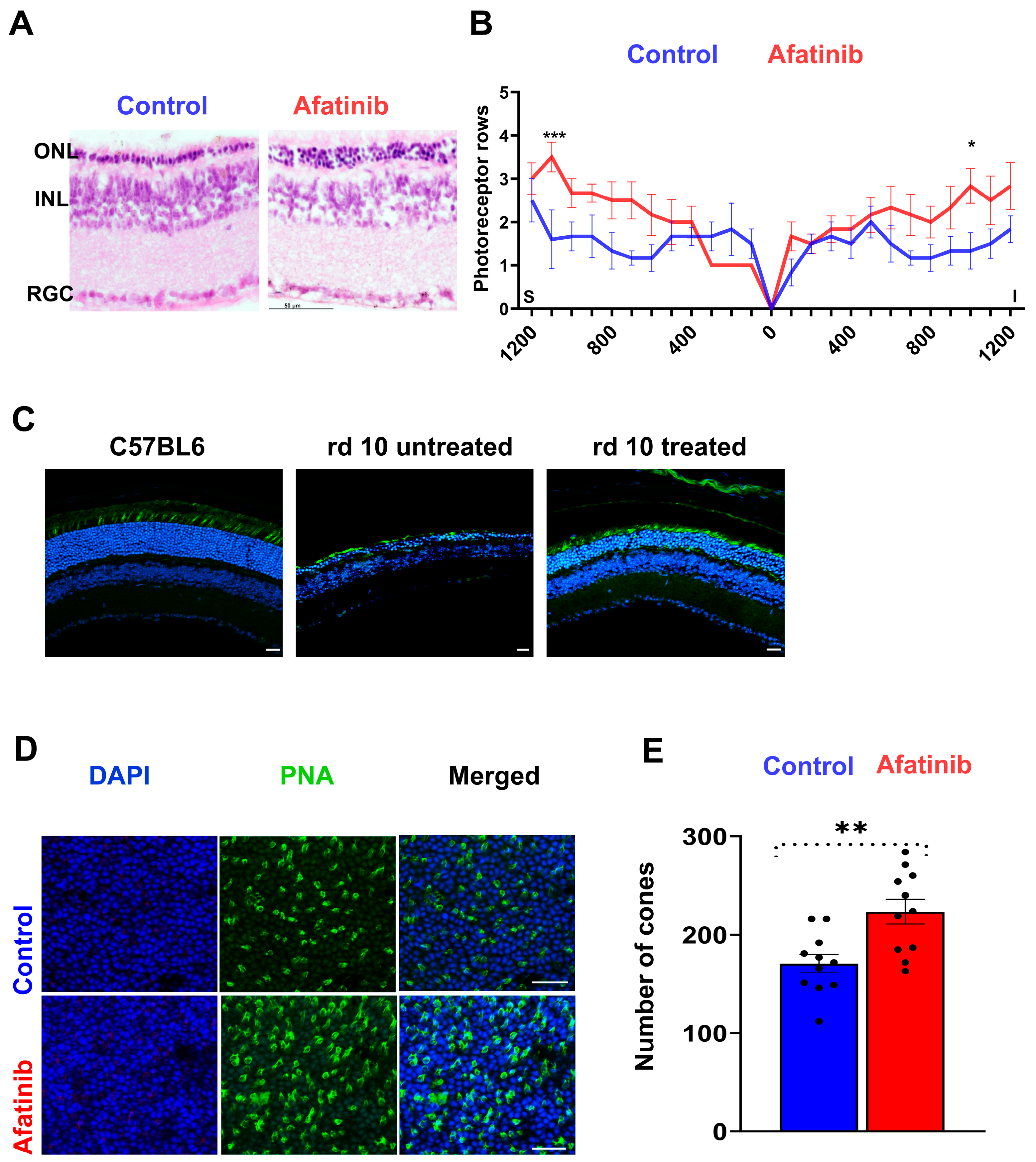
2.3. Treatment of rd10 Mice with Afatinib Reduced TRIB3 and Prevented Retinal Function Loss
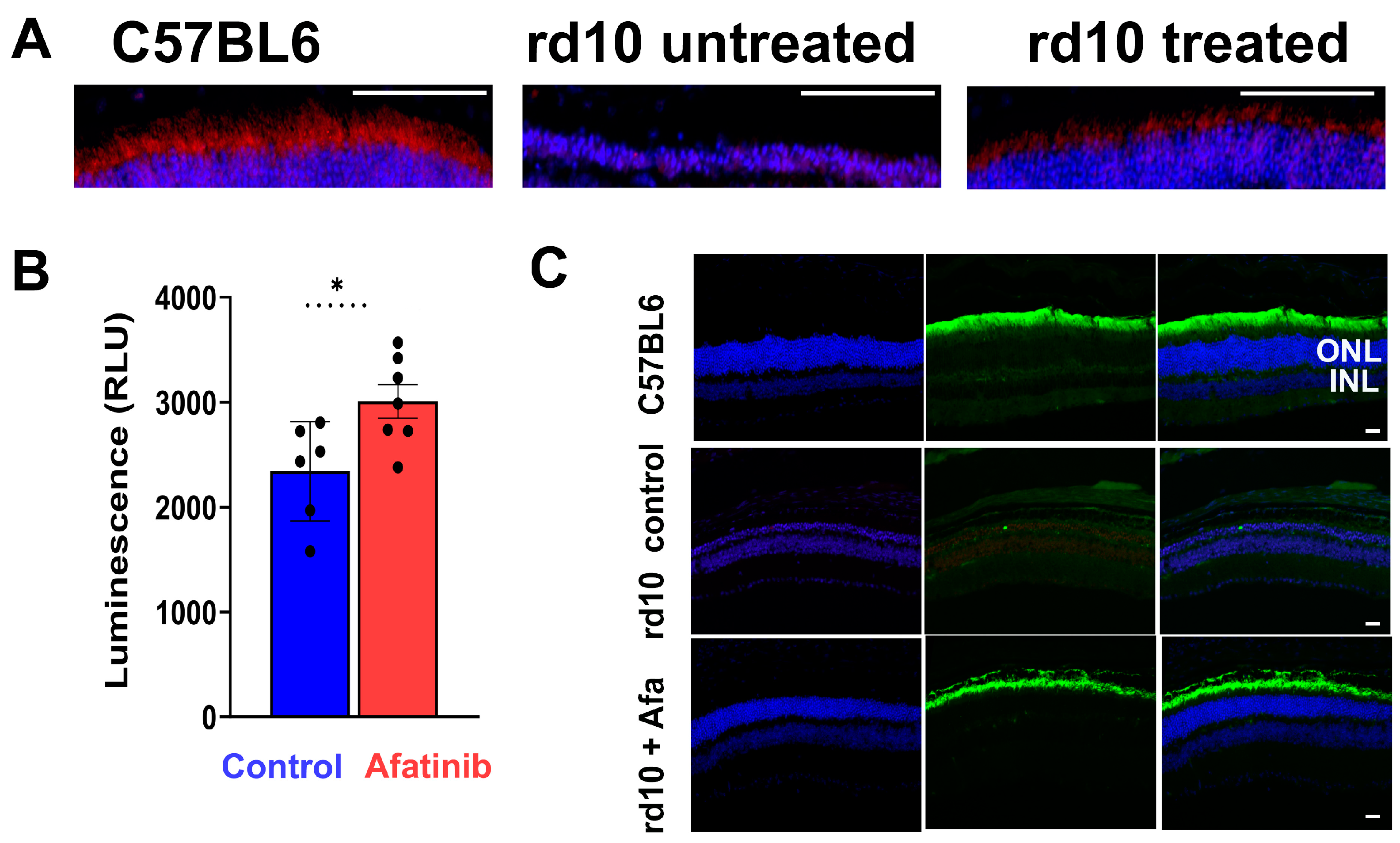
2.4. Treatment of rd10 Mice Prevents Photoreceptor Cell Loss and Increases PDE6β
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Electroretinography (ERG)
4.3. Histology (H&E Staining)
4.4. Immunohistochemistry
4.5. Retinal Explants
4.6. Transfection and Treatment of 661W Cells
4.7. PDE Activity Assay
4.8. Western Blot
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Stefanovska, B.; Andre, F.; Fromigue, O. Tribbles Pseudokinase 3 Regulation and Contribution to Cancer. Cancers 2021, 13, 1822. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.M.; Sun, W.; Wang, Z.H.; Liang, X.; Hua, F.; Li, K.; Lv, X.X.; Zhang, X.W.; Liu, Y.Y.; Yu, J.J.; et al. TRIB3 supports breast cancer stemness by suppressing FOXO1 degradation and enhancing SOX2 transcription. Nat. Commun. 2019, 10, 5720. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Chen, Y.; Wang, L.; Zhang, Y.; Hang, Q.; Li, P.; Zhang, P.; Ji, J.; Song, H.; Chen, M.; et al. PI3K/mTOR inhibitors promote G6PD autophagic degradation and exacerbate oxidative stress damage to radiosensitize small cell lung cancer. Cell Death Dis. 2023, 14, 652. [Google Scholar] [CrossRef] [PubMed]
- Jomary, C.; Cullen, J.; Jones, S.E. Inactivation of the Akt survival pathway during photoreceptor apoptosis in the retinal degeneration mouse. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Starr, C.R.; Pitale, P.M.; Gorbatyuk, M. Translational attenuation and retinal degeneration in mice with an active integrated stress response. Cell Death Dis. 2018, 9, 484. [Google Scholar] [CrossRef] [PubMed]
- Marcal, A.C.; Leonelli, M.; Fiamoncini, J.; Deschamps, F.C.; Rodrigues, M.A.; Curi, R.; Carpinelli, A.R.; Britto, L.R.; Carvalho, C.R. Diet-induced obesity impairs AKT signalling in the retina and causes retinal degeneration. Cell Biochem. Funct. 2013, 31, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.Q.; Huang, W.; Li, K.R.; Liu, Y.Y.; Cao, G.F.; Cao, C.; Jiang, Q. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget 2016, 7, 60123–60132. [Google Scholar] [CrossRef] [PubMed]
- McDougald, D.S.; Papp, T.E.; Zezulin, A.U.; Zhou, S.; Bennett, J. AKT3 Gene Transfer Promotes Anabolic Reprogramming and Photoreceptor Neuroprotection in a Pre-clinical Model of Retinitis Pigmentosa. Mol. Ther. 2019, 27, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Leach, L.L.; Gross, J.M. mTOR activity is essential for retinal pigment epithelium regeneration in zebrafish. PLoS Genet. 2022, 18, e1009628. [Google Scholar] [CrossRef]
- Ma, S.; Venkatesh, A.; Langellotto, F.; Le, Y.Z.; Hall, M.N.; Ruegg, M.A.; Punzo, C. Loss of mTOR signaling affects cone function, cone structure and expression of cone specific proteins without affecting cone survival. Exp. Eye Res. 2015, 135, 1–13. [Google Scholar] [CrossRef]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Justus, S.; Xu, Y.; Pluchenik, T.; Hsu, C.W.; Yang, J.; Duong, J.K.; Lin, C.S.; Jia, Y.; Bassuk, A.G.; et al. Reprogramming towards anabolism impedes degeneration in a preclinical model of retinitis pigmentosa. Hum. Mol. Genet. 2016, 25, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Xiong, G.; Yang, W. Ribosomal protein S6 kinase 1 promotes the survival of photoreceptors in retinitis pigmentosa. Cell Death Dis. 2018, 9, 1141. [Google Scholar] [CrossRef] [PubMed]
- Saltykova, I.V.; Elahi, A.; Pitale, P.M.; Gorbatyuk, O.S.; Athar, M.; Gorbatyuk, M.S. Tribbles homolog 3-mediated targeting the AKT/mTOR axis in mice with retinal degeneration. Cell Death Dis. 2021, 12, 664. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Arrigo, A.; Aragona, E.; Manitto, M.P.; Saladino, A.; Bandello, F.; Battaglia Parodi, M. Gene Therapy in Inherited Retinal Diseases: An Update on Current State of the Art. Front. Med. 2021, 8, 750586. [Google Scholar] [CrossRef] [PubMed]
- Uyama, H.; Mandai, M.; Takahashi, M. Stem-cell-based therapies for retinal degenerative diseases: Current challenges in the establishment of new treatment strategies. Dev. Growth Differ. 2021, 63, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.A.; Bennett, J. The Status of RPE65 Gene Therapy Trials: Safety and Efficacy. Cold Spring Harb Perspect. Med. 2015, 5, a017285. [Google Scholar] [CrossRef] [PubMed]
- Sizova, O.S.; Shinde, V.M.; Lenox, A.R.; Gorbatyuk, M.S. Modulation of cellular signaling pathways in P23H rhodopsin photoreceptors. Cell. Signal. 2014, 26, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, D.M.; Byrne, D.P.; Yeung, W.; Shrestha, S.; Bailey, F.P.; Ferries, S.; Eyers, C.E.; Keeshan, K.; Wells, C.; Drewry, D.H.; et al. Covalent inhibitors of EGFR family protein kinases induce degradation of human Tribbles 2 (TRIB2) pseudokinase in cancer cells. Sci. Signal. 2018, 11, eaat7951. [Google Scholar] [CrossRef]
- Yu, J.J.; Zhou, D.D.; Yang, X.X.; Cui, B.; Tan, F.W.; Wang, J.; Li, K.; Shang, S.; Zhang, C.; Lv, X.X.; et al. TRIB3-EGFR interaction promotes lung cancer progression and defines a therapeutic target. Nat. Commun. 2020, 11, 3660. [Google Scholar] [CrossRef]
- Sundar, J.C.; Munezero, D.; Bryan-Haring, C.; Saravanan, T.; Jacques, A.; Ramamurthy, V. Rhodopsin signaling mediates light-induced photoreceptor cell death in rd10 mice through a transducin-independent mechanism. Hum. Mol. Genet. 2020, 29, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.J.; Dai, X.; Boye, S.E.; Barone, I.; Boye, S.L.; Mao, S.; Everhart, D.; Dinculescu, A.; Liu, L.; Umino, Y.; et al. Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol. Ther. 2011, 19, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Roh, H.; Kim, S.H.; Lee, K.; Im, M.; Oh, S.J. Effective protection of photoreceptors using an inflammation-responsive hydrogel to attenuate outer retinal degeneration. NPJ Regen. Med. 2023, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Tolone, A.; Haq, W.; Fachinger, A.; Roy, A.; Kesh, S.; Rentsch, A.; Wucherpfennig, S.; Zhu, Y.; Groten, J.; Schwede, F.; et al. The PKG Inhibitor CN238 Affords Functional Protection of Photoreceptors and Ganglion Cells against Retinal Degeneration. Int. J. Mol. Sci. 2023, 24, 15277. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Tolone, A.; Paquet-Durand, F.; Welinder, C.; Schwede, F.; Ekstrom, P. The photoreceptor protective cGMP-analog Rp-8-Br-PET-cGMPS interacts with cGMP-interactors PKGI, PDE1, PDE6, and PKAI in the degenerating mouse retina. J. Comp. Neurol. 2023, 531, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, M.; Xiong, G.; Han, X.; Lee, V.W.H.; So, K.F.; Chiu, K.; Xu, Y. Trans-Sclera Electrical Stimulation Improves Retinal Function in a Mouse Model of Retinitis Pigmentosa. Life 2022, 12, 1917. [Google Scholar] [CrossRef]
- Orlans, H.O.; Barnard, A.R.; Patricio, M.I.; McClements, M.E.; MacLaren, R.E. Effect of AAV-Mediated Rhodopsin Gene Augmentation on Retinal Degeneration Caused by the Dominant P23H Rhodopsin Mutation in a Knock-In Murine Model. Hum. Gene Ther. 2020, 31, 730–742. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ung, T.T.; Starr, C.R.; Zhylkibayev, A.; Saltykova, I.; Gorbatyuk, M. Development of TRIB3-Based Therapy as a Gene-Independent Approach to Treat Retinal Degenerative Disorders. Int. J. Mol. Sci. 2024, 25, 4716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094716
Ung TT, Starr CR, Zhylkibayev A, Saltykova I, Gorbatyuk M. Development of TRIB3-Based Therapy as a Gene-Independent Approach to Treat Retinal Degenerative Disorders. International Journal of Molecular Sciences. 2024; 25(9):4716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094716
Chicago/Turabian StyleUng, Trong Thuan, Christopher R. Starr, Assylbek Zhylkibayev, Irina Saltykova, and Marina Gorbatyuk. 2024. "Development of TRIB3-Based Therapy as a Gene-Independent Approach to Treat Retinal Degenerative Disorders" International Journal of Molecular Sciences 25, no. 9: 4716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094716