

Dual Deletion of Keap1 and Rbpjκ Genes in Liver Leads to Hepatomegaly and Hypercholesterolemia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Keap1 Deletion in RbpjF/F::AlbCre Mice Elicits More Severe, Persistent Hepatic Toxicity and Hepatomegaly than Observed in RbpjF/F::AlbCre Mice
2.2. Plasma Clinical Chemistry and Hepatic Lipid Analyses in Keap1F/F::RbpjF/F::AlbCre Mice and Modified Genotypes during the Growth Period
2.3. Altered Expression of Hepatic De Novo Lipid Synthesis Genes at the Transcriptional Level in Early Development among Key Genotypes
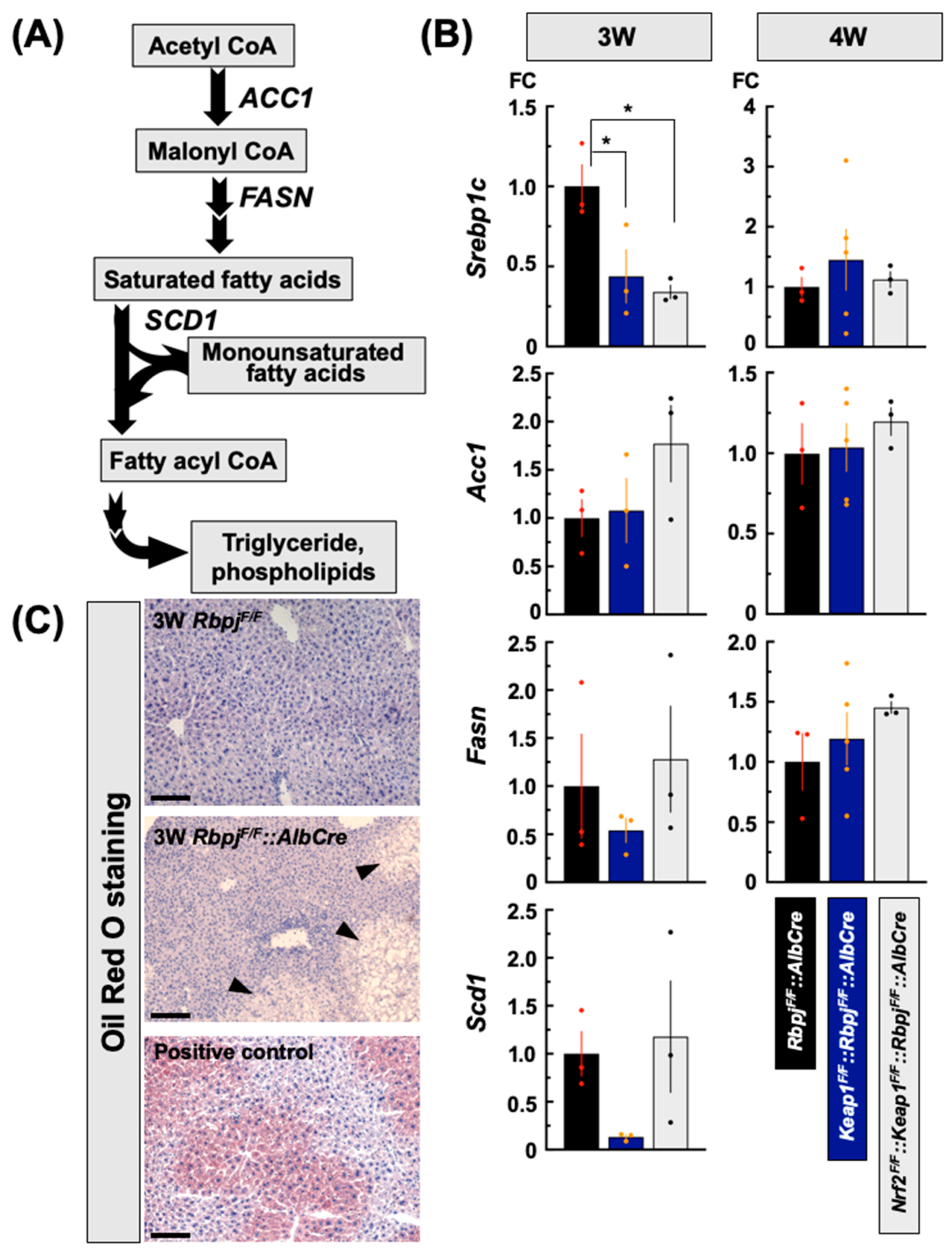
2.3.1. Effect on De Novo Fatty Acid Synthesis-Related Gene Transcripts
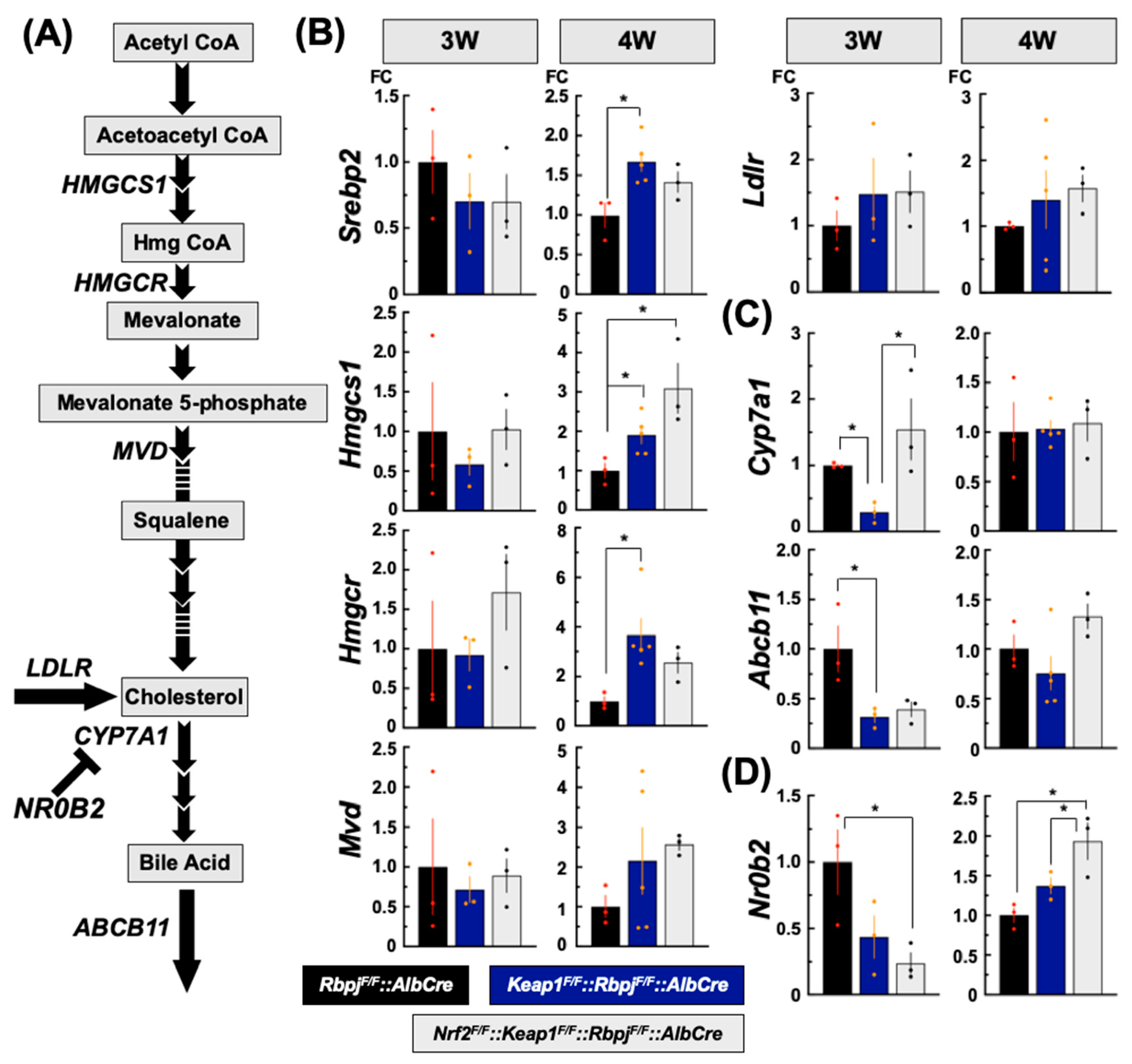
2.3.2. Effect on De Novo Cholesterol Synthesis-Related Gene Transcripts
2.3.3. Effect on Bile Acid Synthesis-Related Gene Transcripts
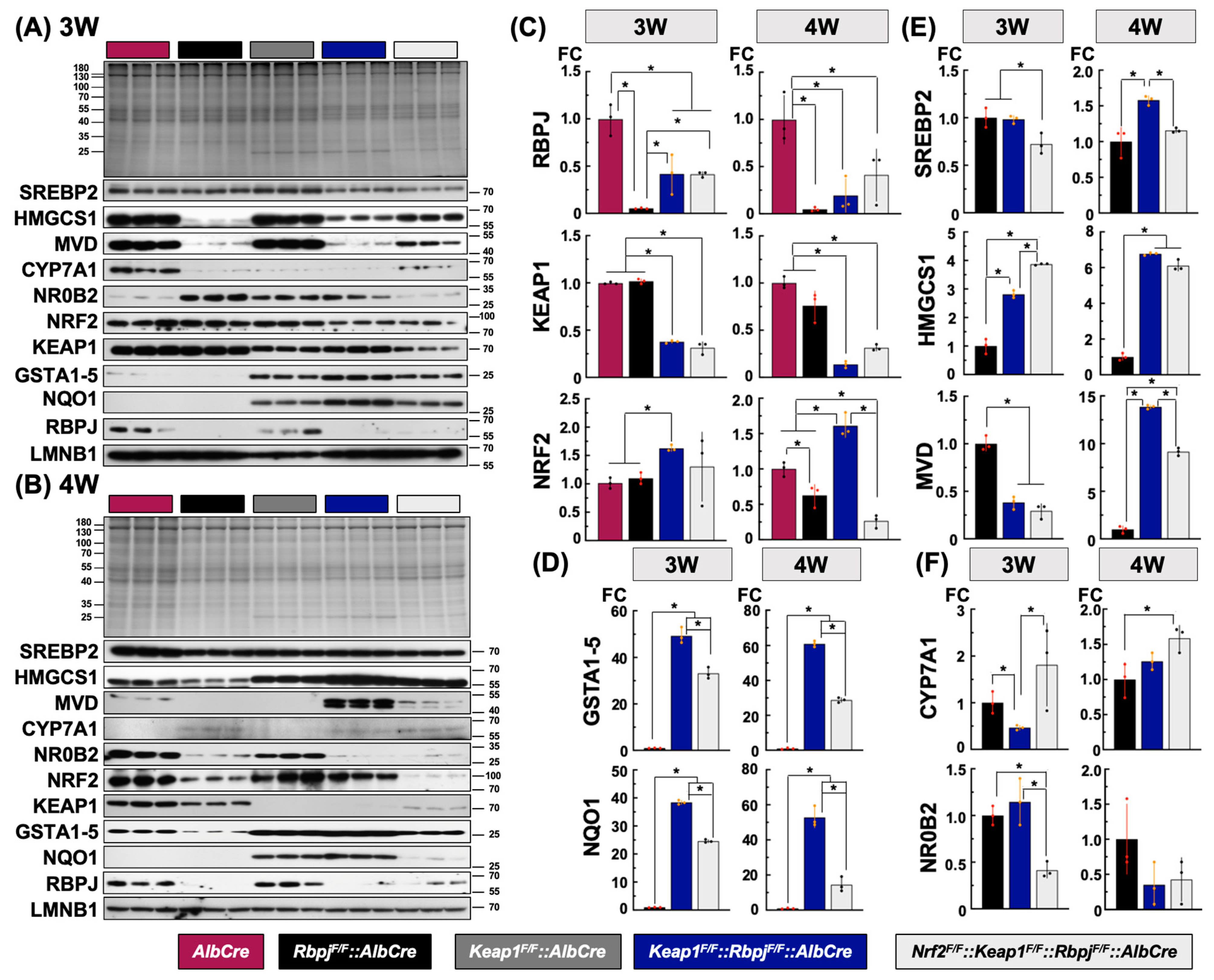
2.4. Protein Level Alterations of Hepatic De Novo Lipid Synthesis-Related Gene Expression during Early Development among Key Genotypes
2.4.1. Protein Expression Related to Hepatic Cholesterol Synthesis among the Cholestasis Genotypes
2.4.2. Protein Expression Related to Hepatic Bile Acid Synthesis among the Cholestasis Genotypes
3. Discussion
3.1. Fatty Acids and Triglycerides Synthetic Pathway
3.2. Cholesterol Synthetic Pathway
3.3. Bile Acid Synthetic Pathway
3.4. How Does Depletion of NOTCH and Enhanced NRF2 Signaling Exacerbate Hyperlipidemia?
4. Materials and Methods
4.1. Animals
4.2. Blood Collection for Biochemical Analyses and Dissection of Liver
4.3. Histology
4.4. Isolation and Purification of Total RNA and RT-PCR
4.5. Hepatic Triglyceride and Cholesterol Assay
4.6. Immune Blotting Analyses
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid. Res. 2013, 52, 175–191. [Google Scholar] [CrossRef]
- Gilbert, M.A.; Bauer, R.C.; Rajagopalan, R.; Grochowski, C.M.; Chao, G.; McEldrew, D.; Nassur, J.A.; Rand, E.B.; Krock, B.L.; Kamath, B.M.; et al. Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum. Mutat. 2019, 40, 2197–2220. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, G.H.; Sokol, R.J.; Stathos, T.H.; Knisely, A.S. Proliferation to paucity: Evolution of bile duct abnormalities in a case of Alagille syndrome. Pediatr. Dev. Pathol. 2001, 4, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kawaichi, M.; Matsunami, N.; Ryo, H.; Nishida, Y.; Honjo, T. The Drosophila RBP-J kappa gene encodes the binding protein for the immunoglobulin J kappa recombination signal sequence. J. Biol. Chem. 1991, 266, 23334–23340. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, Y.; Yamamoto, Y.; Iwanari, H.; Maruyama, S.; Furukawa, T.; Matsunami, N.; Honjo, T. Biochemical and immunological characterization of the DNA binding protein (RBP-J kappa) to mouse J kappa recombination signal sequence. J. Biochem. 1992, 112, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Matsuno, K.; Fortini, M.E. Notch signaling. Science 1995, 268, 225–332. [Google Scholar] [CrossRef] [PubMed]
- Weisend, C.M.; Kundert, J.A.; Suvorova, E.S.; Prigge, J.R.; Schmidt, E.E. Cre activity in fetal albCre mouse hepatocytes: Utility for developmental studies. Genesis 2009, 47, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Magnuson, M.A. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000, 26, 149–150. [Google Scholar] [CrossRef]
- Kellendonk, C.; Opherk, C.; Anlag, K.; Schütz, G.; Tronche, F. Hepatocyte-specific expression of Cre recombinase. Genesis 2000, 26, 151–153. [Google Scholar] [CrossRef]
- Tharehalli, U.; Svinarenko, M.; Kraus, J.M.; Kuhlwein, S.D.; Szekely, R.; Kiesle, U.; Scheffold, A.; Barth, T.F.E.; Kleger, A.; Schirmbeck, R.; et al. YAP Activation Drives Liver Regeneration after Cholestatic Damage Induced by Rbpj Deletion. Int. J. Mol. Sci. 2018, 19, 3801. [Google Scholar] [CrossRef]
- Dědič, T.; Jirsa, M.; Keil, R.; Rygl, M.; Šnajdauf, J.; Kotalová, R. Alagille Syndrome Mimicking Biliary Atresia in Early Infancy. PLoS ONE 2015, 10, e0143939. [Google Scholar] [CrossRef]
- Davenport, M. Biliary atresia: Clinical aspects. Semin Pediatr. Surg. 2012, 21, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Tsuma, Y.; Fukuhara, S.; Kodo, K. A case of Infantile Alagille Syndrome with severe dyslipidemia: NewiInsight into lipid metabolism and therapeutics. J. Endocr. Soc. 2022, 6, bvac005. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.A.; Chen, S.Y.; Gadhvi, G.; Feng, L.; Gromer, K.D.; Abdala-Valencia, H.; Nam, K.; Dominguez, S.T.; Montgomery, A.B.; Reyfman, P.A.; et al. Transcriptional profiling of pediatric cholestatic livers identifies three distinct macrophage populations. PLoS ONE 2021, 16, e0244743. [Google Scholar] [CrossRef] [PubMed]
- Vyas, D.; Baptista, P.M.; Brovold, M.; Moran, E.; Gaston, B.; Booth, C.; Samuel, M.; Atala, A.; Soker, S. Self-assembled liver organoids recapitulate hepatobiliary organogenesis in vitro. Hepatology 2018, 67, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Chartoumpekis, D.V.; Kensler, T.W.; Wakabayashi, N. NRF2 and the Moirai: Life and death decisions on cell fates. Antioxid. Redox Signal. 2023, 38, 684–708. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tabbi-Anneni, I.; Gunda, V.; Wang, L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am. J. Physiol. Gastrointest Liver Physiol. 2010, 299, G1211–G1221. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 2010, 245, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Ziros, P.G.; Psyrogiannis, A.I.; Papavassiliou, A.G.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes 2011, 60, 2465–2473. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Skoko, J.J.; Chartoumpekis, D.V.; Kimura, S.; Slocum, S.L.; Noda, K.; Palliyaguru, D.L.; Fujimuro, M.; Boley, P.A.; Tanaka, Y.; et al. Notch-Nrf2 axis: Regulation of Nrf2 gene expression and cytoprotection by notch signaling. Mol. Cell Biol. 2014, 34, 653–663. [Google Scholar] [CrossRef]
- Sparks, E.E.; Huppert, K.A.; Brown, M.A.; Washington, M.K.; Huppert, S.S. Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 2010, 51, 1391–1400. [Google Scholar] [CrossRef]
- Sparks, E.E.; Perrien, D.S.; Huppert, K.A.; Peterson, T.E.; Huppert, S.S. Defects in hepatic Notch signaling result in disruption of the communicating intrahepatic bile duct network in mice. Dis. Model Mech. 2011, 4, 359–367. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology-divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Kang, S.; Davis, R.A. Cholesterol and hepatic lipoprotein assembly and secretion. Biochim. Biophys. Acta 2000, 1529, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.L.; Strautnieks, S.S.; Jacquemin, E.; Hadchouel, M.; Sokal, E.M.; Hooiveld, G.J.; Koning, J.H.; De Jager-Krikken, A.; Kuipers, F.; Stellaard, F.; et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology 1999, 117, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.A.; Tabor, D.; Kast, H.R.; Venkateswaran, A. Regulation of gene expression by SREBP and SCAP. Biochim. Biophys. Acta 2000, 1529, 103–113. [Google Scholar] [CrossRef]
- Wible, R.S.; Tran, Q.T.; Fathima, S.; Sutter, C.H.; Kensler, T.W.; Sutter, T.R. Pharmacogenomics of Chemically Distinct Classes of Keap1-Nrf2 Activators Identify Common and Unique Gene, Protein, and Pathway Responses In Vivo. Mol. Pharmacol. 2018, 93, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, C.K.; Shah, S.N. Decarboxylation of mevalonate pyrophosphate is one rate-limiting step in hepatic cholesterol synthesis in suckling and weaned rats. Biochem. Biophys. Res. Commun. 1976, 69, 42–47. [Google Scholar] [CrossRef]
- Chen, C.L.; Paul, L.N.; Mermoud, J.C.; Steussy, C.N.; Stauffacher, C.V. Visualizing the enzyme mechanism of mevalonate diphosphate decarboxylase. Nat. Commun. 2020, 11, 3969. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Up to date on cholesterol 7 alpha-hydroxylase (CYP7A1) in bile acid synthesis. Liver Res. 2020, 4, 47–63. [Google Scholar] [CrossRef]
- Kubo, Y.; Beckmann, R.; Fragoulis, A.; Conrads, C.; Pavanram, P.; Nebelung, S.; Wolf, M.; Wruck, C.J.; Jahr, H.; Pufe, T. Nrf2/ARE Signaling Directly Regulates SOX9 to Potentially Alter Age-Dependent Cartilage Degeneration. Antioxidants 2022, 11, 263. [Google Scholar] [CrossRef] [PubMed]
- Walter, T.J.; Vanderpool, C.; Cast, A.E.; Huppert, S.S. Intrahepatic bile duct regeneration in mice does not require Hnf6 or Notch signaling through Rbpj. Am. J. Pathol. 2014, 184, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.S.; Patel, H.; Allaire, N.; Thai, A.; Cullen, P.; Ryan, S.; Lukashev, M.; Bista, P.; Huang, R.; Rhodes, K.J.; et al. Pharmacodynamics of Dimethyl Fumarate Are Tissue Specific and Involve NRF2-Dependent and -Independent Mechanisms. Antioxid. Redox Signal. 2016, 24, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Ziros, P.G.; Zaravinos, A.; Iskrenova, R.P.; Psyrogiannis, A.I.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Hepatic gene expression profiling in Nrf2 knockout mice after long-term high-fat diet-induced obesity. Oxid. Med. Cell Longev. 2013, 2013, 340731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lickteig, A.J.; Liu, J.; Csanaky, I.L.; Klaassen, C.D. Effects of ablation and activation of Nrf2 on bile acid homeostasis in male mice. Toxicol. Appl. Pharmacol. 2020, 403, 115170. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Tang, C.; Cao, P.; Zhou, L.; Chen, X. Metabolic Activation of Retrorsine may Disrupt Bile Acid Homeostasis in Mice through the Nrf2 Pathway. Curr. Drug Metab. 2021, 22, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, O.; Murphy, P.; Prydz, H.; Kolsto, A.B. Interaction of the CNC-bZIP factor TCF11/LCR-F1/Nrf1 with MafG: Binding-site selection and regulation of transcription. Nucleic Acids Res. 1998, 26, 512–520. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 2001, 174, 103–113. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109.e1015. [Google Scholar] [CrossRef] [PubMed]
- Akl, M.G.; Li, L.; Baccetto, R.; Phanse, S.; Zhang, Q.; Trites, M.J.; McDonald, S.; Aoki, H.; Babu, M.; Widenmaier, S.B. Complementary gene regulation by NRF1 and NRF2 protects against hepatic cholesterol overload. Cell Rep. 2023, 42, 112872. [Google Scholar] [CrossRef] [PubMed]
- Tun, T.; Hamaguchi, Y.; Matsunami, N.; Furukawa, T.; Honjo, T.; Kawaichi, M. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 1994, 22, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Wang, X.H.; Hao, L.H.; Wang, H.; Zhang, X.Y.; Muhammad, I.; Qi, Y.; Li, G.L.; Sun, X.Q. Nrf2 is crucial for the down-regulation of Cyp7a1 induced by arachidonic acid in Hepg2 cells. Environ. Toxicol. Pharmacol. 2017, 52, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Garruti, G.; Wang, H.H.; Bonfrate, L.; de Bari, O.; Wang, D.Q.; Portincasa, P. A pleiotropic role for the orphan nuclear receptor small heterodimer partner in lipid homeostasis and metabolic pathways. J. Lipids 2012, 2012, 304292. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Engel, J.D.; Yamamoto, M. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J. Biol. Chem. 2005, 280, 4483–4490. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Ahn, H.; Hagey, L.R.; Romanoski, C.E.; Lee, R.G.; Graham, M.J.; Motohashi, H.; Yamamoto, M.; Edwards, P.A. MAFG is a transcriptional repressor of bile acid synthesis and metabolism. Cell Metab. 2015, 21, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Beltroy, E.P.; Richardson, J.A.; Horton, J.D.; Turley, S.D.; Dietschy, J.M. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology 2005, 42, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Landis, C.S.; Jin, G.Y.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated with Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Sozen, E.; Demirel-Yalciner, T.; Sari, D.; Ozer, N.K. Cholesterol accumulation in hepatocytes mediates IRE1/p38 branch of endoplasmic reticulum stress to promote nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2022, 191, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, J.; Zhao, K.; Gao, L.; Zhao, J. Cholesterol-induced toxicity: An integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021, 33, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.M.; Potteti, H.R.; Mariani, T.J.; Biswal, S.; Reddy, S.P. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am. J. Respir Cell Mol. Biol. 2011, 45, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tanigaki, K.; Yamamoto, N.; Kuroda, K.; Yoshimoto, M.; Nakahata, T.; Ikuta, K.; Honjo, T. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int. Immunol. 2002, 14, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Yagishita, Y.; Joshi, T.; Kensler, T.W. Forced Hepatic Expression of NRF2 or NQO1 Impedes Hepatocyte Lipid Accumulation in a Lipodystrophy Mouse Model. Int. J. Mol. Sci. 2023, 24, 13345. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E180–E195. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Yagishita, Y.; Fazzari, M.; Palliyaguru, D.L.; Rao, U.N.; Zaravinos, A.; Khoo, N.K.; Schopfer, F.J.; Weiss, K.R.; Michalopoulos, G.K.; et al. Nrf2 prevents Notch-induced insulin resistance and tumorigenesis in mice. JCI Insight 2018, 3, e97735. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wakabayashi, N.; Yagishita, Y.; Joshi, T.; Kensler, T.W. Dual Deletion of Keap1 and Rbpjκ Genes in Liver Leads to Hepatomegaly and Hypercholesterolemia. Int. J. Mol. Sci. 2024, 25, 4712. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094712
Wakabayashi N, Yagishita Y, Joshi T, Kensler TW. Dual Deletion of Keap1 and Rbpjκ Genes in Liver Leads to Hepatomegaly and Hypercholesterolemia. International Journal of Molecular Sciences. 2024; 25(9):4712. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094712
Chicago/Turabian StyleWakabayashi, Nobunao, Yoko Yagishita, Tanvi Joshi, and Thomas W. Kensler. 2024. "Dual Deletion of Keap1 and Rbpjκ Genes in Liver Leads to Hepatomegaly and Hypercholesterolemia" International Journal of Molecular Sciences 25, no. 9: 4712. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094712