

Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species

1
CAS Key Laboratory of Science and Technology on Applied Catalysis, iChEM, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
2
University of Chinese Academy of Sciences, Beijing 100049, China
*
Author to whom correspondence should be addressed.
Molecules 2024, 29(9), 1987; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091987
Submission received: 14 March 2024
/
Revised: 18 April 2024
/
Accepted: 22 April 2024
/
Published: 26 April 2024
(This article belongs to the Topic Catalysis: Homogeneous and Heterogeneous, 2nd Edition)
Abstract
:Reppe carbonylation of acetylene is an atom-economic and non-petroleum approach to synthesize acrylic acid and acrylate esters, which are key intermediates in the textile, leather finishing, and polymer industries. In the present work, a noble metal-free Co@SiO2 catalyst was prepared and evaluated in the methoxycarbonylation reaction of acetylene. It was discovered that pretreatment of the catalyst by different reductants (i.e., C2H2, CO, H2, and syngas) greatly improved the catalytic activity, of which Co/SiO2-H2 demonstrated the best performance under conditions of 160 °C, 0.05 MPa C2H2, 4 MPa CO, and 1 h, affording a production rate of 4.38 gMA+MP gcat−1 h−1 for methyl acrylate (MA) and methyl propionate (MP) and 0.91 gDMS gcat−1 h−1 for dimethyl succinate (DMS), respectively. Transmission electron microscopy (TEM), X-ray diffraction (XRD), and diffuse reflectance infrared Fourier transform spectra of CO adsorption (CO-DRIFTS) measurements revealed that an H2 reduction decreased the size of the Co nanoparticles and promoted the formation of hollow architectures, leading to an increase in the metal surface area and CO adsorption on the catalyst. The hot filtration experiment confirmed that Co2(CO)8 was generated in situ during the reaction or at the pre-activation stage, which served as the genuine active species. Our work provides a facile and convenient approach to the in situ synthetization of Co2(CO)8 for a Reppe carbonylation reaction.
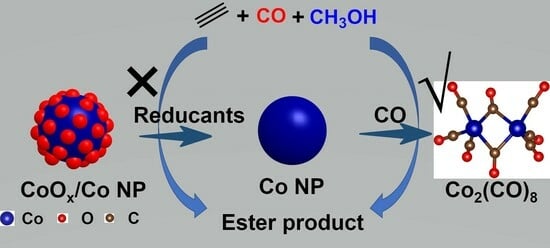
1. Introduction
Reppe carbonylation reactions of acetylene (including hydrocarboxylation, alkoxycarbonylation, etc.) (Scheme 1) provide atom-economic and non-petroleum routes to produce acrylic acid (AA), methyl acrylate, and dimethyl succinate [1,2], which are raw materials and key intermediates that are widely used in the textile, leather finishing, and polymer industries, with a global demand of 10 million tons annually [3,4,5].
Various catalysts have been developed for the carbonylation reactions of acetylene, of which Ni-based catalysts are the original ones [6]. Reppe and coworkers were the first to discover that Ni(CO)4 was able to catalyze the hydrocarboxylation of acetylene to produce AA under 3 MPa CO and acetylene (1/1, v/v) [2]. Following this, other nickel catalysts, such as (copper-promoted) nickel halides [7,8], nickel acetates [9], and Ni-P complexes [10], were also reported. Among others, Badische Anilin-und-Soda-Fabrik (BASF) developed the commercial catalytic system NiBr2-CuBr2-CH3SOOOH for hydrocarbonylation of acetylene [11], which, however, suffers from a difficulty in the separation of catalyst products [12]. To tackle this problem, metal oxides (such as SiO2, Al2O3, vermiculite, and MCM-41) [13,14,15] and zeolites (e.g., NaY and ZSM-5) [16] were applied as support materials of Ni. However, these catalysts have one or more drawbacks, which include a short catalyst lifetime, harsh reaction conditions, and metal leaching. For example, Shi and coworkers reported that Ni/Y served as an efficient catalyst for the hydrocarboxylation of acetylene [17], and the AA space-time yield reached 62 gacrylic acid gcat−1 h−1 under reaction conditions of 235 °C and 3.6 MPa. Although they claimed that the catalysis followed a heterogeneous pathway, the Ni/Y material suffered from severe coking. Yan and coworkers prepared a NiO/AlOOH catalyst, which gave an AA space-time yield as high as 412 gacrylic acid gcat−1 h−1, yet it was discovered that the leached Ni-carbonyls were the true active species [12]. Given the high toxicity and instability of Ni(CO)4, which might generate in situ during a reaction, the potential application of Ni-based catalysts is limited.
Other catalysts based on group VIII transition metals, such as Pd, Ir, and Ru, have also been explored [18]. For instance, Drent and coworkers developed a Pd complex with the ligand of 2-pyridylphosphine (2-PyPPh2) [19], which exhibited excellent catalytic performance in the methoxycarbonylation of propyne, and the TOF reached as high as 40,000 h−1 with 99.95% selectivity to methyl methacrylate. The 2-pyridyl ring was found to be crucial for the catalytic activity, which, if replaced by a phenyl or 4-pyridyl group, led to a drastic decrease in TOF. Nonetheless, strong acid promoters, e.g., p-toluenesulfonic acid, are indispensable for the reaction, which would cause serious environmental issues [20]. Ding and coworkers developed porous organic polymer-supported single-site Pd catalysts, in which the 2-pyridylphosphine ligand and p-toluenesulfonic acid were incorporated into the framework of the support [21,22], and the methoxycarbonylation reaction of acetylene was allowed to run in the absence of an extra acid promoter. However, this approach suffered from tedious procedures for the preparation of the support [23,24]. In addition, the high cost and low abundance of Pd compromised its efficiency.
Cobalt is also the metal of choice for Reppe carbonylation reactions due to its earth abundance and high catalytic activity [25,26,27]. Pyridine-promoted cobalt carbonyls demonstrated high efficiency in converting butadiene to adipic acid in a pilot-scale process developed by BASF [28,29]. Cobalt carbonyls were also competent catalysts in the alkoxycarbonylation reactions of alkenes [30]. Nevertheless, to the best of our knowledge, few reports have been produced on the Co-catalyzed methoxycarbonylation of acetylene. In the present work, we prepared SiO2-supported Co catalysts and investigated their catalytic performance in the methoxycarbonylation of acetylene in the absence of acid promoters. It was discovered that the pre-activation atmosphere (i.e., H2, C2H2, syngas, CO, and NH3) had a profound impact on the catalytic activity, which increased following the trend of C2H2 < CO < syngas ≈ H2. Under reaction conditions of 160 °C and 0.05 MPa C2H2 and 4 MPa CO, Co/SiO2 exhibited high catalytic activity, leading to a production rate of 4.2 gMA+MP gcat−1 h−1 for MA and MP and 0. 9 gDMS gcat−1 h−1 for DMS, respectively. Mechanism studies by XRD and hot filtration test revealed that Co nanoparticles underwent dynamic evolution during the reaction, where cobalt carbonyls were formed in situ and served as the genuine active species. Our work provided an approach for the in situ generation of catalytically active species for the methoxycarbonylation reaction, thus circumventing the employment of costly and unstable conventional Co2(CO)8 catalysts, which are generally synthesized under harsh conditions of high temperatures and high pressures [31,32,33].
2. Results and Discussion
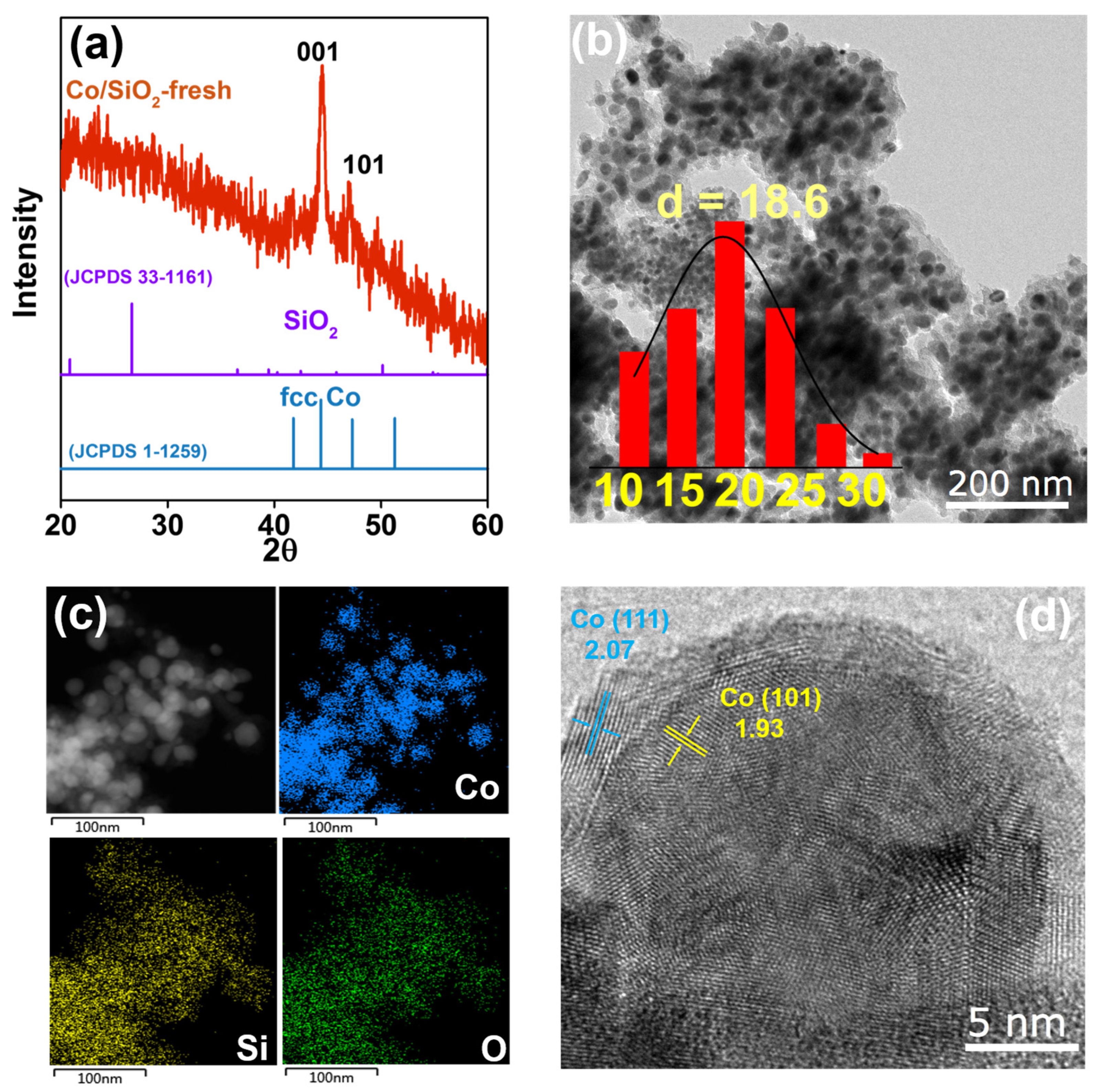
The Co/SiO2 catalysts were prepared by the one-pot synthetic method, followed by calcination and reduction [34]. The XRD patterns (Figure 1a) exhibited characteristic diffraction peaks of metallic Co with an fcc structure (JCPDS 00-01-1259), and the size of the Co was calculated to be 28 nm according to the Scherrer equation [35]. No diffraction peaks ascribed to SiO2 (JCPDS 00-33-1161) were present, probably owing to the amorphous nature of SiO2 in the sample. The TEM images (Figure 1b) and energy-dispersive X-ray (EDX) spectra (Figure 1c) showed that Co nanoparticles were homogeneously distributed on SiO2, most of which had an average size of 18.6 nm. However, the examination of different regions showed that there were also smaller Co nanoparticles with sizes of 1–2 nm (Figure S1). The lattice spacing was measured to be 2.07 nm and 1.93 nm (Figure 1d), corresponding to the (111) and (101) plane in fcc Co, respectively, which were in agreement with the XRD measurements.
The as-prepared Co/SiO2 catalysts were evaluated in the methoxycarbonylation reaction of acetylene under conditions of 0.05 MPa C2H2, 4 MPa CO, 5 mL CH3OH, and 160 °C. However, no products were detected in the three catalysts with different Co loadings after a 1 h reaction (Table 1, entries 1–3). It is assumed that the passive layer of CoOx outside of the metallic Co might suppresses the reactant to access the catalytically active sites [36]; therefore, 1 MPa H2 was charged into the reactor to activate the catalyst in situ. As expected, under this condition, the Co/SiO2 samples demonstrated high catalytic performance for the target reaction. Three main products were detected (i.e., MA, MP, and DMS) after a 1 h reaction (Table 1, entries 4–6), of which MA and DMS were produced by the methoxycarbonylation and dimethoxycarbonylation of acetylene, respectively, while MP was derived from the hydrogenation of MA. Specifically, in the 41.7% Co/SiO2 catalyst (Table 1, entry 4), all the MA had been completely hydrogenated to MP, which was produced with a rate as high as 4.42gMP gcat−1 h−1, and DMS was yielded with a relatively lower rate of 0. 58 gDMS gcat−1 h−1. The other two Co/SiO2 catalysts with different Co loadings were also tested in the reaction. The 31.6% Co/SiO2 sample showed similar catalytic performance to the 41.7% Co/SiO2 sample (Table 1, entry 5). However, the catalytic activity of the 25.6% Co/SiO2 sample, with an even lower Co loading, decreased drastically (Table 1, entry 6), and only the methoxycarbonylation product was yielded, with a rate of 0.39 gMA+MP gcat−1 h−1. The reason might be that with a decrease in Co loading, the size of the Co became smaller, and in turn, the interaction between Co and SiO2 became stronger (e.g., cobalt silicate might be formed) [34,37], leading to a more difficult reduction of the CoOx species. The other support-material-loaded Co catalysts were also evaluated in the reaction. However, Co/WOx, Co/HAP, Co/TiO2, and Co/S-1 were totally inactive for the reaction (Table 1, entries 7–10). Metallic Co powder was inefficient for the reaction as well (Table 1, entry 11).
As the pre-activation of Co/SiO2 by H2 improved the catalytic activity remarkably, the other reductants, i.e., C2H2, CO, syngas, and NH3, were also employed to pre-activate the sample to see if the catalytic activity could be further improved. Except for NH3, the pre-activation was conducted in an autoclave in CH3OH under different reductant gases at 160 °C for 1 h, and after that, the autoclave was flushed with N2 and recharged with the reactant gas (0.05 MPa C2H2 and 4 MPa CO), and the reaction was allowed to run at 160 °C for 1 h. The pretreatment by NH3 was carried out according to the literature [38]. As shown in Table 2, the pretreatment under Ar made no contribution to the activity (Table 2, entry 1), thus precluding the possibility that CH3OH served as the reductant. By contrast, the pre-activations under C2H2, CO, or syngas atmosphere all led to obvious yields of the three products, and the production rate increased following the trend of Co/SiO2-C2H2 (Table 2, entry 2) < Co/SiO2-CO (entry 3) < Co/SiO2-syngas (entry 5) ≈ Co/SiO2-H2 (entry 4). The improvement in catalytic activity by pretreatment with C2H2 or CO was surprising, as no products were detected using the as-made Co/SiO2 catalyst under 0.05 MPa C2H2 and 4 MPa CO at 160 °C for 1 h, as mentioned before (Table 1, entry 1). We suppose there might be an induction period of >1 h; accordingly, the reaction was once again conducted using an as-made Co/SiO2 catalyst under 0.05 MPa C2H2 and 4 MPa CO for a long time (i.e., 2 h); however, no products were detected. On the contrary, the Co/SiO2-NH3 sample showed no catalytic activity for the reaction (Table 2, entry 6). The reason for this might be that NH3 adsorbed strongly on the surface of metallic Co, and thereby, the adsorption of the reactants on the catalyst was prevented. To verify this, we carried out a control experiment, where the methoxycarbonylation of acetylene was conducted under conditions of 0.1 MPa NH3, 0.05 MPa C2H2, 4 MPa CO, 5 mL CH3OH, 160 °C, and 1 h using Co2(CO)8 as the catalyst (Co2(CO)8 was used here because it has been proven to be the genuine active species in the following section). It was found that no product was yielded, suggesting that the Co2(CO)8 catalyst was poisoned by NH3.
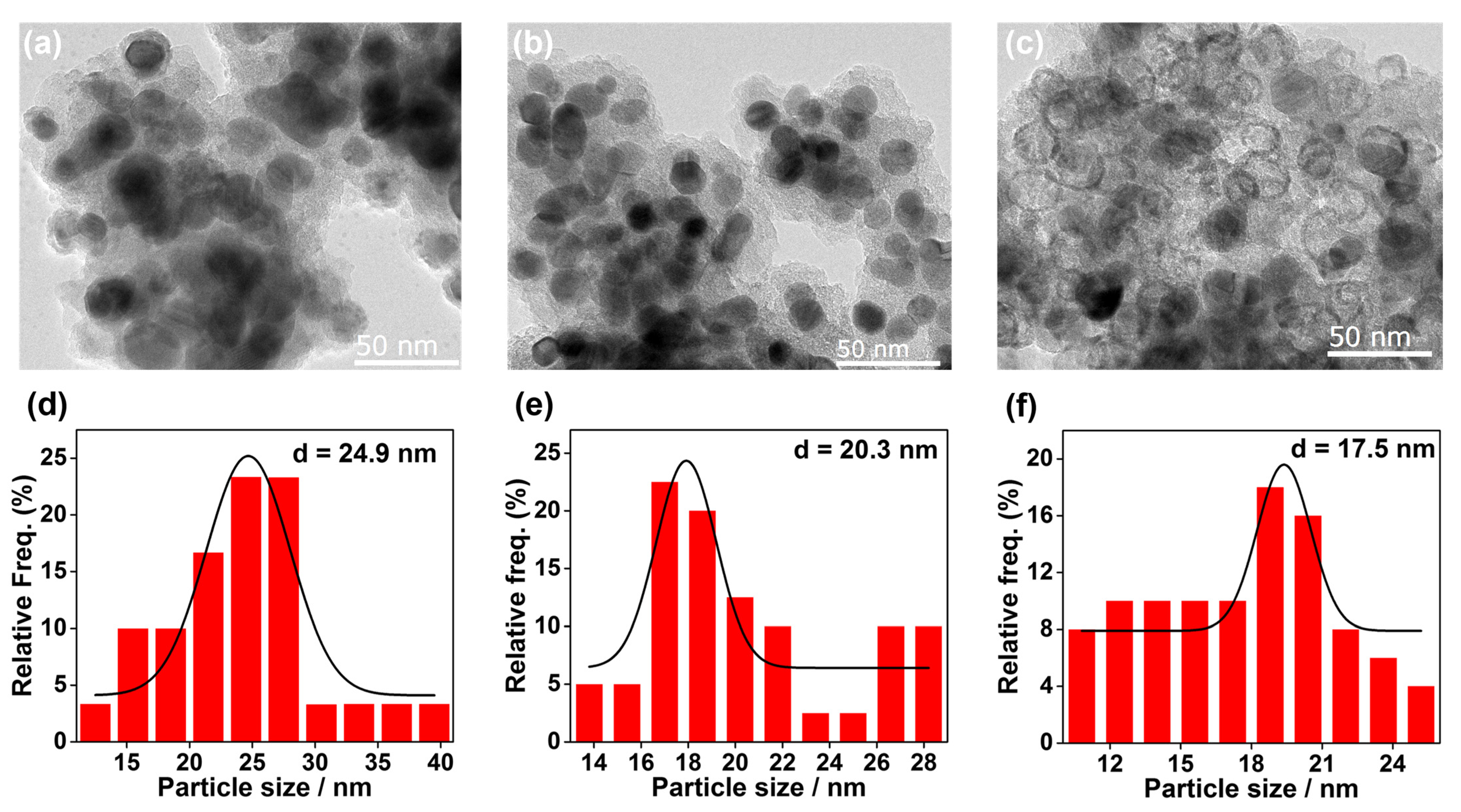
On the basis of the above results, it is inferred that pretreatment with a distinct reducing gas should modify the structure of the Co/SiO2 catalyst, where the active species are more prone to be formed. Accordingly, TEM, XRD, and CO-DRIFT measurements of the activated catalysts were conducted. Figure 2 shows the TEM images and particle size histograms for different pre-activated Co/SiO2 samples. The average particle size of metallic Co was 24.9 nm, and it was 20.3 nm for Co/SiO2-C2H2 (Figure 2a,d) and Co/SiO2-CO (Figure 2b,e), respectively. Compared with fresh Co/SiO2 (18.6 nm), it is obvious that the C2H2 and CO pre-activation induce an increase in particle size. In sharp contrast, in the Co/SiO2-H2 sample (Figure 2c,f and Figure S2), not only had the size of the Co decreased to 17.5 nm, but also, a lot of Co nanoparticles had been reconstructed to hollow shells. This reconstruction during H2 reduction might result from the strain induced by a variation in the metal–metal distances between the CoOx surface layer and the inner metallic Co [39].
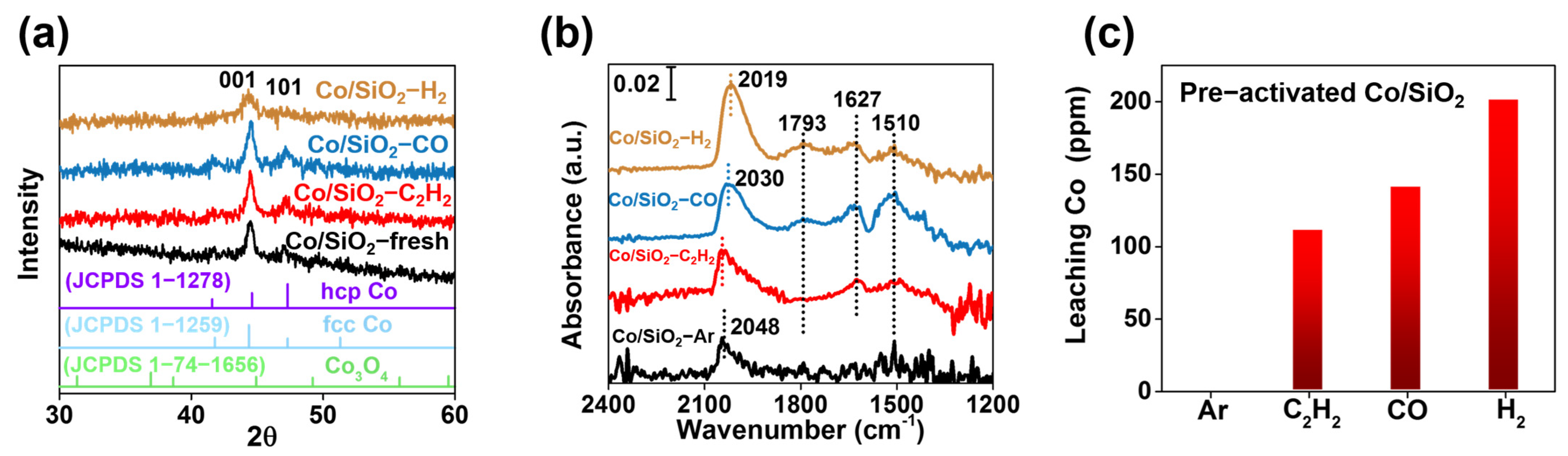
As shown in Figure 3a, the intensity of the diffraction peaks at 44.4° and 47.3°, attributed to the (111) and (101) plane of metallic Co [40] in the samples of Co/SiO2-C2H2 and Co/SiO2-CO, increased greatly compared with those of the fresh Co/SiO2, indicating an increase in the size of the Co nanoparticles. In sharp contrast, for the Co/SiO2-H2 sample, the diffraction peaks attenuated instead, suggesting a decrease in the Co particle size, or a reduction in the crystallinity of the metallic Co. These results were consistent with the TEM examination results but quite different from conventional experimental results, where H2 reduction generally results in the sintering of metal nanoparticles [41]. CO-DRIFTs were also carried out to study the electronic properties of Co. As shown in Figure 3b, four adsorption bands appeared on different samples. The band at 2048 cm−1 was ascribed to the ν(C≡O) of CO molecules that were linearly adsorbed on metallic Co [42], while the one at 1793 cm−1 was assigned to the ν(C≡O) of CO molecules in multi-bonded carbonyls on Co [43]. The other two bands at 1627 and 1510 cm−1 were attributed to the νasymm(C=O) of bicarbonate species and νasymm(C=O) of carbonate species, respectively [44,45]. It was found that the integrated area of the CO band at 2048 cm−1 and 1793 cm−1 increased with the trend of Co/SiO2-C2H2 < Co/SiO2-CO < Co/SiO2-H2, indicating an increased surface area of the metallic Co, which should come from the reduction of the passive layer of CoOx. In addition, this peak red-shifted to 2030 cm−1 on Co/SiO2-CO and further to 2019 cm−1 on Co/SiO2-H2, which, according to the TEM images, might result from the increased proportion of corner and edge sites in the Co nanoparticles with a decrease in sizes.
Stability is a key criterion for judging the quality of a catalyst [46]. Therefore, we also studied the recyclability of the Co/SiO2 catalyst. When the used catalyst was separated from the reaction mixture by filtration, it was found that the filtrate was dark red in color, suggesting severe leaching of Co [47,48]. The concentration of Co in the filtrate of different samples increased, following the trend of Co/SiO2-C2H2 < Co/SiO2-CO < Co/SiO2-H2 (Figure 3c), which was in agreement with that of the catalytic activity. In addition, the Co/SiO2 catalyst was discovered to already undergo Co leaching after the pretreatment with the reductant. In particular, for the Co/SiO2–syngas sample, after the pre-activation using syngas, the solid sample was separated by filtration and was then subjected to a batch of reactions (Figure S4); however, no products were yielded at all. By contrast, when the dark-red filtrate (containing 200 ppm Co) was employed as a catalyst, production rates of 3.76 gMA+MP gcat−1 h−1 of MA and MP and 0.49 gDMS gcat−1 h−1 of DMS were achieved (Table 3), which was quite similar to that of the Co/SiO2–syngas (Table 2, entry 5). These results indicated that it was the leached Co species that contributed to the overall catalytic activity. 13C NMR measurement was then conducted to identify the leached Co species; however, the filtrate did not show any signal because of the paramagnetic nature of Co. The yellow color of the filtrate implied that the leached Co species might be Co2(CO)8. To confirm this point, we used commercial Co2(CO)8 as a catalyst for the methoxycarbonylation of acetylene. It was discovered that Co2(CO)8 demonstrated similar catalytic activity and product distribution to the filtrate (Figure 4), verifying that Co2(CO)8 was the true active species for the reaction.
Co2(CO)8 is known as an efficient catalyst for the alkoxycarbonylation and hydroformylation of alkenes [49,50,51], although no reports on its catalytic performance in the methoxycarbonylation of acetylene have been published. Our work thus demonstrated that Co2(CO)8 is also a competent catalyst for this transformation to produce MA and MP. Traditionally, Co2(CO)8 is quite unstable, and its synthesis requires either harsh reaction conditions (i.e., 140–230 °C, 10–70 MPa) or stoichiometric reducing reagents (such as metal powders, NaBH4, or Na2S2SO3) or phosphine oxide promoters. Li and coworkers recently reported that Co2(CO)8 could be formed in situ from Co/MoS2 under conditions of 6 MPa CO, 140 °C, and 15 h, and MoS2 was claimed to be critical for this process [28]. This work demonstrated that when using conventional SiO2 as a support material, Co2(CO)8 could also be generated in situ under conditions of 4 MPa CO, 1 MPa H2, 160 °C, and 1 h, thus providing a facile, straightforward approach to prepare Co2(CO)8 for Reppe carbonylation reactions.
3. Materials and Methods
The Co/SiO2 was prepared according to the literature [34]. Briefly, 2.91 g Co(NO3)2·6H2O (10 mmol) and 2.08 g TEOS (10 mmol) were dissolved in 50 mL mixed liquor of water and ethanol (3/1, v/v) and stirred for 10 min. After, 5 mL of NH3·H2O was added to the above solution, and the suspension was stirred at room temperature for another 8 h. The precipitate was separated and collected by filtration, washed with deionized water, and dried at 100 °C overnight. The obtained solid was calcined at 500 °C in a muffle furnace for 4 h and was then reduced in flowing pure hydrogen (100 mL/min) for 3 h at 600 °C. The obtained Co/SiO2 catalyst was labeled as 41.7% Co/SiO2. The other two catalysts with different Co loadings (i.e., 31.6% Co/SiO2 and 25.6% Co/SiO2) were prepared using similar procedures, except for changing the amount of TEOS to 15 and 20 mmol, respectively.
The other support material (i.e., WOx, hydroxyapatite (HAP), and TiO2)-loaded Co catalysts were synthesized by the impregnation method. First, 5 mL Co(NO3)2·6H2O solution (10 mg Co/mL) and 5 mL ultrapure water were mixed and stirred to form a transparent solution, and then, 1 g carrier was added to the above solution. After the mixture was stirred at room temperature for 24 h, the excess water was removed by rotary evaporation until dry. The resulting power was further dried at 373 K overnight and then reduced in flowing pure hydrogen (100 mL/min) for 3 h at 600 °C.
The preparation of the Co2(CO)8 solution in methanol from Co/SiO2 started with adding 10 mg of fresh Co/SiO2 to a 30 mL autoclave with 5 mL methanol, and then, the autoclave was purged with N2 repeatedly (4 times) to remove gaseous and dissolved oxygen. After that, the autoclave was charged with 4 MPa CO and 1 MPa H2, and the mixture was stirred (600 rpm) at 160 °C for 4 h. Then, the autoclave was placed into an ice bath until the temperature was below 5 °C. The solid was separated by filtration, and the yellow filter liquor was collected and kept at 0 C.
Other comparison materials, including Co power and Co2(CO)8, are commercial samples at analytically pure levels.
3.1. Characterization
The concentration of Co in the filtrate after pretreatment or reaction was determined by inductively coupled plasma spectroscopy (ICP-AES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corporation, Waltham, MA, USA). The patterns of XRD were recorded on a PANalytical X’ pert diffractometer with a Cu-Kα radiation source, operated at 40 kV and 40 mA under a continuous mode in the 2θ range of 10°~90°. The morphology and element distribution of samples were observed by STEM and EDS experiments, which were performed on a JEM-2100F Transmission Electron Microscope (JEOL, Singapore) with a spatial resolution of 0.19 nm at 20 kV, equipped with the Oxford Instruments ISIS/INCA EDS system with an Oxford Pentafet Ultrathin Window (UTW) detector. Before the microscopy examination, the sample was demagnetization-treated using a magnet. After that, the sample was ultrasonically dispersed in ethanol for 5–10 min, and then, a drop of the suspension was dropped on a copper TEM grid coated with a thin holey carbon film. CO-DRIFTS (diffuse reflectance infrared Fourier transform spectroscopy) experiments were performed by means of a Bruker Equinox70 spectrometer (Bruker, Singapore), equipped with a mercury–cadmium–telluride detector at a resolution of 4 cm−1, using 16 scans in a range of 400–4000 cm−1. Prior to the measurement, the catalysts were pretreated in situ with H2 (30 mL/min) at 160 °C for 30 min, and then, the flow was switched to He (30 mL/min) at 170 °C for another 30 min to remove surface H. After that, the catalyst cooled down to room temperature, and background spectra were recorded at 20 °C in He flow.
3.2. Catalyst Evaluation
The acetylene methoxycarbonylation (AMC) reaction was performed in a 30 mL autoclave equipped with a quartz lining. The autoclave was filled with 10 mg of a catalyst and 5 mL methanol, and then, the autoclave was purged with N2 repeatedly (4 times) to remove gaseous and dissolved oxygen. After that, it was charged with 1 MPa 5% C2H2-95% He, 1 MPa H2, and 4 MPa CO. The reaction was started by heating the mixture to 160 °C under vigorous stirring at a speed of 600 rpm. After the reaction, the reactor was cooled to room temperature, and the liquid product was analyzed using n-pentanol as the internal by gas chromatography (Agilent 7890B, Santa Clara, CA, USA), equipped with an HP-INNO WAX column (30 m × 320 μm × 0.25 μm). Only methyl acrylate (MA), methyl propionate (MP), dimethyl succinate (DMS), 1,1-dimethoxypropane (2,2-DMP), and 3-pentanone were detected in the liquid product. The reaction selectivity was calculated based on the molar ratio of MA, MP, and DMS to the total liquid product, and the reaction rate was calculated by the total mass of MA, MP, and DMS produced per mass of Co per hour. The ‘others’ in the result are the total mass of 3-pentanone and 2,2-DMP. It should be emphasized when using Co2(CO)8 as a catalyst before the reactant gas is charged that the autoclave should be placed into an ice bath until the temperature is below 5 °C; otherwise, the dissolved Co2(CO)8 will be swept away owing the volatile property of Co2(CO)8.
The pretreatment of the Co/SiO2 catalyst and the catalytic evaluation were conducted in a methoxycarbonylation reaction of acetylene. Taking Co/SiO2-C2H2 as an example, 10 mg of fresh Co/SiO2 was added into a 30 mL autoclave with 5 mL methanol, and then, the autoclave was purged with N2 repeatedly (4 times) to remove the air and dissolved oxygen. After that, the autoclave was charged with 5% C2H2–95% He until 1 MPa. The autoclave was heated at 160 °C for 1 h with vigorous stirring at a speed of 600 rpm. When the reactor was cooled to room temperature, the gas in the reactor was evacuated, and the autoclave was recharged with C2H2 (1 MPa) and CO (4 MPa) for acetylene methoxycarbonylation. A similar pretreatment process was also conducted using 1 MPa H2, 4 MPa CO, or syngas (a mixture of 1 MPa H2 and 4 MPa CO).
4. Conclusions
In summary, a noble metal-free Co@SiO2 catalyst was prepared and tested in the methoxycarbonylation reaction of acetylene. The pre-activation of the catalyst using different reductant gases (i.e., C2H2, CO, H2, and syngas) was found to be crucial for the high catalytic performance. The catalytic activity increased following the trend of Co/SiO2-C2H2 < Co/SiO2-CO < Co/SiO2–syngas ≈ Co/SiO2-H2, and high production rates of 4.38 gMA+MP gcat−1 h−1 and 0.91 gDMS gcat−1 h−1 were obtained for Co/SiO2-H2 under conditions of 160 °C, 0.05 MPa C2H2, 4 MPa CO, and 1 h. The characterization by TEM, XRD, and CO-DRIFTS revealed that the size or crystallinity of Co nanoparticles was greatly decreased upon pre-reduction by H2, which remarkably increased the metal surface area and CO adsorption on the catalyst. The hot filtration experiment and controlled measurement confirmed that Co was leached into the solution in the form of Co2(CO)8 and served as the genuine active species. Our work provides a facile and convenient approach to synthesize Co2(CO)8 for a Reppe carbonylation reaction, thus avoiding the direct usage of highly costly and unstable Co2(CO)8 as a catalyst.
Supplementary Materials
The following supporting information can be downloaded at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/molecules29091987/s1: Figure S1: STEM images of Co/SiO2 at different magnifications; Figure S2: (a–c) TEM images of pre-activated Co/SiO2-H2; Figure S3: DRIFT spectra of CO adsorption on different samples; Figure S4: Diagram illustration of separation process.
Author Contributions
Conceptualization, A.W. (Aiqin Wang) and L.Z.; methodology, A.W. (An Wang); investigation, A.W. (An Wang) and H.C.; writing—original draft preparation, A.W. (An Wang); writing—review and editing, L.Z.; supervision, A.W. (Aiqing Wang). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key R&D Program of China (2023YFA1506603), the NSFC Center for Single-atom Catalysis (Grant NO. 22388102), National Natural Science Foundation of China (22132006, 22172159), the CAS Project for Young Scientists in Basic Research (YSBR-022), and the Youth Innovation Promotion Association CAS (2022185).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kiss, G. Palladium-catalyzed Reppe carbonylation. Chem. Rev. 2001, 101, 3435–3456. [Google Scholar] [CrossRef]
- Trotus, I.T.; Zimmermann, T.; Schueth, F. Catalytic Reactions of Acetylene: A Feedstock for the Chemical Industry Revisited. Chem. Rev. 2014, 114, 1761–1782. [Google Scholar] [CrossRef] [PubMed]
- Kalck, P.; Urrutigoïty, M. Recent improvements in the alkoxycarbonylation reaction catalyzed by transition metal complexes. Inorg. Chim. Acta 2015, 431, 110–121. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. Chemicals from Alkynes with Palladium Catalysts. Chem. Rev. 2014, 114, 1783–1826. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Wang, J.Q.; Yuan, Q.; Song, X.E.; Mu, J.L.; Wei, Y.; Yan, L.; Sun, F.F.; Feng, S.Q.; Cai, Y.T.; et al. Palladium and Ruthenium Dual-Single-Atom Sites on Porous Ionic Polymers for Acetylene Dialkoxycarbonylation: Synergetic Effects Stabilize the Active Site and Increase CO Adsorption. Angew. Chem. Int. Ed. 2023, 62, 202307570. [Google Scholar] [CrossRef] [PubMed]
- Reppe, W. New developments in the field of chemical acetylene and coal. Experientia 1949, 5, 93–110. [Google Scholar] [CrossRef]
- Bhattacharyya, S.K.; Sen, A.K. Catalytic Syntheses of Acrylic Acid and Ethyl Acrylate from Acetylene, Carbon Monoxide, and Water or Ethanol under Pressure. Ind. Eng. Chem. Process Des. Dev. 1964, 3, 169–176. [Google Scholar] [CrossRef]
- Bhattacharyya, S.K.; Nag, S.N. Catalytic synthesis of ethyl propionate from ethylene, carbon monoxide and ethanol at high pressure. J. Appl. Chem. 1962, 12, 182. [Google Scholar] [CrossRef]
- Cui, L.; Yang, X.G.; Zeng, Y.; Chen, Y.T.; Wang, G.Y. A unique nickel-base nitrogen-oxygen bidentate ligand catalyst for carbonylation of acetylene to acrylic acid. Mol. Catal. 2019, 468, 57–61. [Google Scholar] [CrossRef]
- Tang, C.M.; Zeng, Y.; Cao, P.; Yang, X.G.; Wang, G.Y. The Nickel and Copper-Catalyzed Hydroformylation of Acetylene with Carbon Monoxide to Acrylic Acid. Catal. Lett. 2009, 129, 189–193. [Google Scholar] [CrossRef]
- Lin, T.J.; Meng, X.; Shi, L. Catalytic hydrocarboxylation of acetylene to acrylic acid using Ni2O3 and cupric bromide as combined catalysts. J. Mol. Catal. A Chem. 2015, 396, 77–83. [Google Scholar] [CrossRef]
- Li, Y.K.; Yan, L.F.; Zhang, Q.F.; Yan, B.H.; Cheng, Y. A recyclable heterogeneous-homogeneous-heterogeneous NiO/AlOOH catalysis system for hydrocarboxylation of acetylene to acrylic acid. RSC Adv. 2020, 10, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.K.; Bhattach, D.P. Catalytic synthesis of n-propyl acrylate from acetylene carbon monoxide and n-propanol under pressure. J. Appl. Chem. 1966, 16, 18. [Google Scholar] [CrossRef]
- Hu, G.; Guo, D.; Shang, H.J.; Sun, Y.K.; Zeng, J.M.; Li, J.B.; Zhu, M.Y. Expanded Two-Dimensional Layered Vermiculite Supported Nickel Oxide Nanoparticles Provides High Activity for Acetylene Carbonylation to Synthesize Acrylic Acid. Catal. Lett. 2020, 150, 674–682. [Google Scholar] [CrossRef]
- Hu, G.; Guo, D.; Shang, H.J.; Sun, Y.K.; Zeng, J.M.; Li, J.B. Microwave-Assisted Rapid Preparation of Vermiculite-Loaded Nano-Nickel Oxide As a Highly Efficient Catalyst for Acetylene Carbonylation to Synthesize Acrylic Acid. Chem. Select. 2020, 5, 2940–2948. [Google Scholar] [CrossRef]
- Xie, H.; Lin, T.J.; Shi, L.; Meng, X. Acetylene carbonylation over Ni-containing catalysts: Role of surface structure and active site distribution. RSC Adv. 2016, 6, 97285–97292. [Google Scholar] [CrossRef]
- Lin, T.J.; Meng, X.; Shi, L. Ni-exchanged Y-zeolite. An efficient heterogeneous catalyst for acetylene hydrocarboxylation. Appl. Catal. A 2014, 485, 163–171. [Google Scholar] [CrossRef]
- Wu, X.F.; Fang, X.J.; Wu, L.P.; Jackstell, R.; Neumann, H.; Beller, M. Transition-Metal-Catalyzed Carbonylation Reactions of Olefins and Alkynes: A Personal Account. Acc. Chem. Res. 2014, 47, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Arnoldy, P.; Budzelaar, P.H.M. Homogeneous catalysis by cationic palladium complexes precision catalysis in the carbonylation of alkynes. J. Organomet. Chem. 1994, 475, 57–63. [Google Scholar] [CrossRef]
- Wei, X.M.; Ma, Z.W.; Mu, X.Y.; Lu, J.Z.; Hu, B. Catalyst in Acetylene Carbonylation: From Homogeneous to Heterogeneous. Prog. Chem. 2021, 33, 243–253. [Google Scholar] [CrossRef]
- Chen, X.K.; Zhu, H.J.; Wang, W.L.; Du, H.; Wang, T.; Yan, L.; Hu, X.P.; Ding, Y.J. Multifunctional Single-Site Catalysts for Alkoxycarbonylation of Terminal Alkynes. ChemSusChem 2016, 9, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.K.; Zhu, H.J.; Wang, T.; Li, C.Y.; Yan, L.; Jiang, M.; Liu, J.; Sun, X.P.; Jiang, Z.; Ding, Y.J. The 2V-P,N polymer supported palladium catalyst for methoxycarbonylation of acetylene. J. Mol. Catal. A Chem. 2016, 414, 37–46. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, L.L.; Yu, Z.A.; Zhang, S.X.; Li, L.; Ren, Y.J.; Yang, J.; Liu, X.Y.; Liu, W.; Yang, X.F.; et al. Ethylene Methoxycarbonylation over Heterogeneous Pt1/MoS2 Single-Atom Catalyst: Metal-Support Concerted Catalysis. J. Am. Chem. Soc. 2023, 146, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Feng, S.Q.; Hemberger, P.; Bodi, A.; Song, X.G.; Yuan, Q.; Mu, J.L.; Li, B.; Jiang, Z.; Ding, Y.J. Iodide-Coordinated Single-Site Pd Catalysts for Alkyne Dialkoxycarbonylation. ACS Catal. 2021, 11, 9242–9251. [Google Scholar] [CrossRef]
- Sarkar, B.R.; Chaudhari, R.V. Carbonylation of alkynes, alkenes and alcohols using metal complex catalysts. Catal. Surv. Asia 2005, 9, 193–205. [Google Scholar] [CrossRef]
- Zhang, X.H.; Shen, C.R.; Xia, C.G.; Tian, X.X.; He, L. Alkoxycarbonylation of olefins with carbon dioxide by a reusable heterobimetallic ruthenium-cobalt catalytic system. Green Chem. 2018, 20, 5533–5539. [Google Scholar] [CrossRef]
- Pesa, F.; Haase, T. The cobalt-catalyzed hydroesterification of acrylonitrile. J. Mol. Catal. 1983, 18, 237–249. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhou, H.; Deng, L.; Jia, X.; Li, Y. Molybdenum disulfide promoted co-catalyzed alkoxycarbonylation. J. Catal. 2024, 430, 115349. [Google Scholar] [CrossRef]
- Hofmann, P.; Kosswig, K.; Schaefer, W. Hydrocarboxymethylation—An attractive route from olefins to fatty-acid esters? Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 330–334. [Google Scholar] [CrossRef]
- Huang, W.H.; Jackstell, R.; Spannenberg, A.; Beller, M. An improved cobalt-catalysed alkoxycarbonylation of olefins using secondary phosphine oxide promotors. Catal. Sci. Technol. 2023, 13, 2475–2479. [Google Scholar] [CrossRef]
- Orchin, M. Hydrogenation of organic compounds with synthesis gas. Adv. Catal. 1953, 5, 385–415. [Google Scholar] [CrossRef]
- Satyanarayana, N.; Periasamy, M. Carbonylation of benzyl halides using CoCl2/NaBH4/CO/NaOH reagent system. Tetrahedron Lett. 1987, 28, 2633–2636. [Google Scholar] [CrossRef]
- Itoh, K.; Miura, M.; Nomura, M. Normal pressure double carbonylation of aryl halides using Cobalt(ii) chloride in the presence of either sodium sulfide or sodium-borohydride. Bull. Chem. Soc. Jpn. 1988, 61, 4151–4152. [Google Scholar] [CrossRef]
- Wang, L.X.; Guan, E.J.; Wang, Y.Q.; Wang, L.; Gong, Z.M.; Cui, Y.; Meng, X.J.; Gates, B.C.; Xiao, F.S. Silica accelerates the selective hydrogenation of CO2 to methanol on cobalt catalysts. Nat. Commun. 2020, 11, 14817. [Google Scholar] [CrossRef] [PubMed]
- Muniz, F.T.L.; Miranda, M.A.R.; dos Santos, C.M.; Sasaki, J.M. The Scherrer equation and the dynamical theory of X-ray diffraction. Acta Crystallogr. Sect. A Found. Adv. 2016, 72, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Kaneti, Y.V.; Zhang, J.; He, Y.B.; Wang, Z.J.; Tanaka, S.; Hossain, M.S.A.; Pan, Z.Z.; Xiang, B.; Yang, Q.H.; Yamauchi, Y. Fabrication of an MOF-derived heteroatom-doped Co/CoO/carbon hybrid with superior sodium storage performance for sodium-ion batteries. J. Mater. Chem. 2017, 5, 15356–15366. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Deng, W.P.; Wang, Y. Recent advances in understanding the key catalyst factors for Fischer-Tropsch synthesis. J. Energy Chem. 2013, 22, 27–38. [Google Scholar] [CrossRef]
- Guan, D.Q.; Zhong, J.; Xu, H.Y.; Huang, Y.C.; Hu, Z.W.; Chen, B.; Zhang, Y.; Ni, M.; Xu, X.M.; Zhou, W.; et al. A universal chemical-induced tensile strain tuning strategy to boost oxygen-evolving electrocatalysis on perovskite oxides. Appl. Phys. Res. 2022, 9, 011422. [Google Scholar] [CrossRef]
- LaGrow, A.P.; Lloyd, D.C.; Gai, P.L.; Boyes, E.D. In Situ Scanning Transmission Electron Microscopy of Ni Nanoparticle Redispersion via the Reduction of Hollow NiO. Chem. Mater. 2018, 30, 197–203. [Google Scholar] [CrossRef]
- Du, H.; Jiang, M.; Zhu, H.J.; Huang, C.D.; Zhao, Z.; Dong, W.D.; Lu, W.; Liu, T.; Zhang, Z.C.; Ding, Y.J. Constructing efficient hcp-Co active sites for Fischer-Tropsch reaction on an activated carbon supported cobalt catalyst via multistep activation processes. Fuel 2021, 292, 120244. [Google Scholar] [CrossRef]
- Parastaev, A.; Muravev, V.; Osta, E.H.; van Hoof, A.J.F.; Kimpel, T.F.; Kosinov, N.; Hensen, E.J.M. Boosting CO2 hydrogenation via size-dependent metal-support interactions in cobalt/ceria-based catalysts. Nat. Catal. 2020, 3, 526–533. [Google Scholar] [CrossRef]
- Parastaev, A.; Muravev, V.; Osta, E.H.; Kimpel, T.F.; Simons, J.F.M.; van Hoof, A.J.F.; Uslamin, E.; Zhang, L.; Struijs, J.J.C.; Burueva, D.B.; et al. Breaking structure sensitivity in CO2 hydrogenation by tuning metal-oxide interfaces in supported cobalt nanoparticles. Nat. Catal. 2022, 5, 1051–1060. [Google Scholar] [CrossRef]
- Weststrate, C.J.; van de Loosdrecht, J.; Niemantsverdriet, J.W. Spectroscopic insights into cobalt-catalyzed Fischer-Tropsch synthesis: A review of the carbon monoxide interaction with single crystalline surfaces of cobalt. J. Catal. 2016, 342, 1–16. [Google Scholar] [CrossRef]
- Zhu, H.Q.; Qin, Z.F.; Shan, W.J.; Shen, W.J.; Wang, J.G. Pd/CeO2-TiO2 catalyst for CO oxidation at low temperature:: A TPR study with H2 and CO as reducing agents. J. Catal. 2004, 225, 267–277. [Google Scholar] [CrossRef]
- Tabakova, T.; Boccuzzi, F.B.; Manzoli, M.; Andreeva, D. FTIR study of low-temperature water-gas shift reaction on gold/ceria catalyst. Appl. Catal. A 2003, 252, 385–397. [Google Scholar] [CrossRef]
- Banham, D.; Ye, S.; Pei, K.; Ozaki, J.; Kishimoto, T.; Imashiro, Y. A review of the stability and durability of non-precious metal catalysts for the oxygen reduction reaction in proton exchange membrane fuel cells. J. Power Sources 2015, 285, 334–348. [Google Scholar] [CrossRef]
- Hertrich, M.F.; Scharnagl, F.K.; Pews-Davtyan, A.; Kreyenschulte, C.R.; Lund, H.; Bartling, S.; Jackstell, R.; Beller, M. Supported Cobalt Nanoparticles for Hydroformylation Reactions. Chem. Eur. J. 2019, 25, 5534–5538. [Google Scholar] [CrossRef]
- Zhao, J.J.; He, Y.R.; Wang, F.; Zheng, W.T.; Huo, C.F.; Liu, X.; Jiao, H.J.; Yang, Y.; Li, Y.W.; Wen, X.D. Suppressing Metal Leaching in a Supported Co/SiO2 Catalyst with Effective Protectants in the Hydroformylation Reaction. ACS Catal. 2020, 10, 914–920. [Google Scholar] [CrossRef]
- Rush, L.E.; Pringle, P.G.; Harvey, J.N. Computational Kinetics of Cobalt-Catalyzed Alkene Hydroformylation. Angew. Chem. Int. Ed. 2014, 53, 8672–8676. [Google Scholar] [CrossRef]
- Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Hood, D.M.; Johnson, R.A.; Carpenter, A.E.; Younker, J.M.; Vinyard, D.J.; Stanley, G.G. Highly active cationic cobalt(II) hydroformylation catalysts. Science 2020, 367, 542–549. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Reppe carbonylation reactions of acetylene.

Figure 1.
(a) The XRD patterns of the fresh Co/SiO2 catalyst and the references of fcc Co and SiO2; (b) the transmission electron microscope (TEM) images of Co/SiO2 and the size distribution of Co nanoparticles; (c) the EDX spectra of Co, Si, and O; (d) the TEM image of Co/SiO2 and exposed lattice plane.
Figure 1.
(a) The XRD patterns of the fresh Co/SiO2 catalyst and the references of fcc Co and SiO2; (b) the transmission electron microscope (TEM) images of Co/SiO2 and the size distribution of Co nanoparticles; (c) the EDX spectra of Co, Si, and O; (d) the TEM image of Co/SiO2 and exposed lattice plane.

Figure 2.
Representative TEM images and corresponding particle size histograms for pre-activated catalysts: (a,d) Co/SiO2-C2H2; (b,e) Co/SiO2-CO; (c,f) Co/SiO2-H2.
Figure 2.
Representative TEM images and corresponding particle size histograms for pre-activated catalysts: (a,d) Co/SiO2-C2H2; (b,e) Co/SiO2-CO; (c,f) Co/SiO2-H2.

Figure 3.
(a) XRD patterns of fresh and pre-activated Co/SiO2, as well as reference Co samples; (b) DRIFT spectra of CO adsorption on different samples; (c) amount of leaching Co in reaction filtrate from pre-activated Co/SiO2 (determined by ICP-OES).
Figure 3.
(a) XRD patterns of fresh and pre-activated Co/SiO2, as well as reference Co samples; (b) DRIFT spectra of CO adsorption on different samples; (c) amount of leaching Co in reaction filtrate from pre-activated Co/SiO2 (determined by ICP-OES).

Figure 4.
Catalytic performances in methoxycarbonylation of acetylene using Co leaching and Co2(CO)8. Reaction conditions: 2.89 mg Co2(CO)8, 0.05 MPa C2H4, 4 MPa CO, 5 mL CH3OH,160 °C, 1 h; for filtrate Co, there are 200 ppm Co in solution.
Figure 4.
Catalytic performances in methoxycarbonylation of acetylene using Co leaching and Co2(CO)8. Reaction conditions: 2.89 mg Co2(CO)8, 0.05 MPa C2H4, 4 MPa CO, 5 mL CH3OH,160 °C, 1 h; for filtrate Co, there are 200 ppm Co in solution.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Catalytic performances of various catalysts in methoxycarbonylation reaction of acetylene a.
Table 1.
Catalytic performances of various catalysts in methoxycarbonylation reaction of acetylene a.
| Entry | Catalyst | Product Rate (gproduct gcat−1 h−1) | Yield % | |||
|---|---|---|---|---|---|---|
| MA | MP | DMS | Others | |||
| 1 | a 41.7% Co/SiO2 | 0 | 0 | 0 | 0 | 0 |
| 2 | a 31.6% Co/SiO2 | 0 | 0 | 0 | 0 | 0 |
| 3 | a 25.6% Co/SiO2 | 0 | 0 | 0 | 0 | 0 |
| 4 | b 41.7% Co/SiO2 | 0 | 4.42 | 0.58 | 0.49 | 64.1 |
| 5 | b 31.6% Co/SiO2 | 0.43 | 4.0 | 0.56 | 0.39 | 58.6 |
| 6 | b 25.6% Co/SiO2 | 0.16 | 0.23 | 0 | 0 | ≈0 |
| 7 | b 5% Co/WOx | 0 | 0 | 0 | 0 | 0 |
| 8 | b 5% Co/HAP | 0 | 0 | 0 | 0 | 0 |
| 9 | b 5% Co/TiO2 | 0 | 0 | 0 | 0 | 0 |
| 10 | b Co/S-1 | 0 | 0 | 0 | 0 | 0 |
| 11 | b Co powder | 0 | 0 | 0 | 0 | 0 |
a Reaction conditions: 10 mg catalyst, 0.05 MPa C2H2, 4 MPa CO, 5 mL CH3OH,160 °C, 1 h. b 0.05 MPa C2H2, 4 MPa CO, 1 MPa H2, 5 mL CH3OH,160 °C, 1 h. Others include 1,1-dimethoxypropane (2,2-DMP) and 3-pentanone. Yield was calculated on the basis of the total amount of MA, MP, and DMS.
Table 2.
Catalytic performances of various pre-activated Co/SiO2 in methoxycarbonylation reaction of acetylene a.
Table 2.
Catalytic performances of various pre-activated Co/SiO2 in methoxycarbonylation reaction of acetylene a.
| Entry | Pre-Activation | Product Rate (gproduct gcat−1 h−1) | Reaction Rate (molMA+MP+DMS molCo h−1) | ||
|---|---|---|---|---|---|
| MA | MP | DMS | |||
| 1 | 1 MPa Ar | 0 | 0 | 0 | 0 |
| 2 | 0.05 MPa C2H2 | 1.51 | 0 | 0.42 | 0.31 |
| 3 | 4 MPa CO | 2.91 | 0.03 | 0.54 | 0.56 |
| 4 | 1 MPa H2 | 4.23 | 0.15 | 0.91 | 0.85 |
| 5 | 1 MPa H2 + 4 MPa CO | 3.4 | 0.16 | 1.25 | 0.77 |
| 6 | 0.01 MPa NH3 | 0 | 0 | 0 | 0 |
a Reaction conditions: 10 mg catalyst, 0.05 MPa C2H2, 4 MPa CO, 5 mL CH3OH, 160 °C, 1 h.
Table 3.
Catalytic performances of filtrate and residual solids in methoxycarbonylation reaction of acetylene a.
Table 3.
Catalytic performances of filtrate and residual solids in methoxycarbonylation reaction of acetylene a.
| Catalyst | Product Rate (gproduct gcat−1 h−1) | ||
|---|---|---|---|
| MA | MP | DMS | |
| a Filtrate | 3.6 | 0.16 | 0.35 |
| a Residual solid | 0 | 0 | 0 |
a Reaction conditions: 10 mg catalyst, 0.05 MPa C2H2, 4 MPa CO, 5 mL CH3OH, 160 °C, 1 h.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, A.; Cao, H.; Zhang, L.; Wang, A. Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species. Molecules 2024, 29, 1987. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091987
AMA Style
Wang A, Cao H, Zhang L, Wang A. Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species. Molecules. 2024; 29(9):1987. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091987
Chicago/Turabian StyleWang, An, Hongchen Cao, Leilei Zhang, and Aiqin Wang. 2024. "Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species" Molecules 29, no. 9: 1987. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091987