
Benefits of Combined Fluorescence Lifetime Imaging Microscopy and Fluorescence Correlation Spectroscopy for Biomedical Studies Demonstrated by Using a Liposome Model System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material and Sample Preparation
2.2. Fluorescence Measurements
2.3. 2P Fluorescence Excitation Spectra
2.4. 2P-FLIM Recordings
2.5. Fluorescence Correlation Spectroscopy
3. Results and Discussion
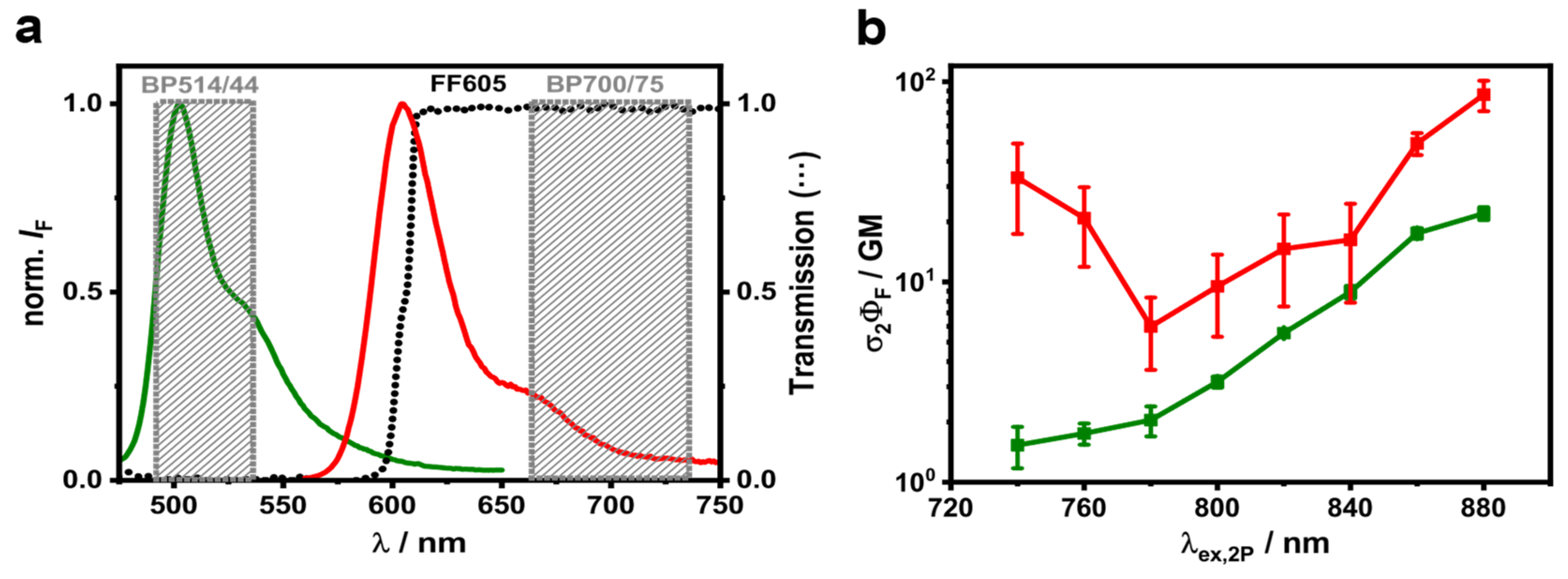
3.1. 2P Fluorescence Excitation Spectra of Fluorescently Labelled LUVs
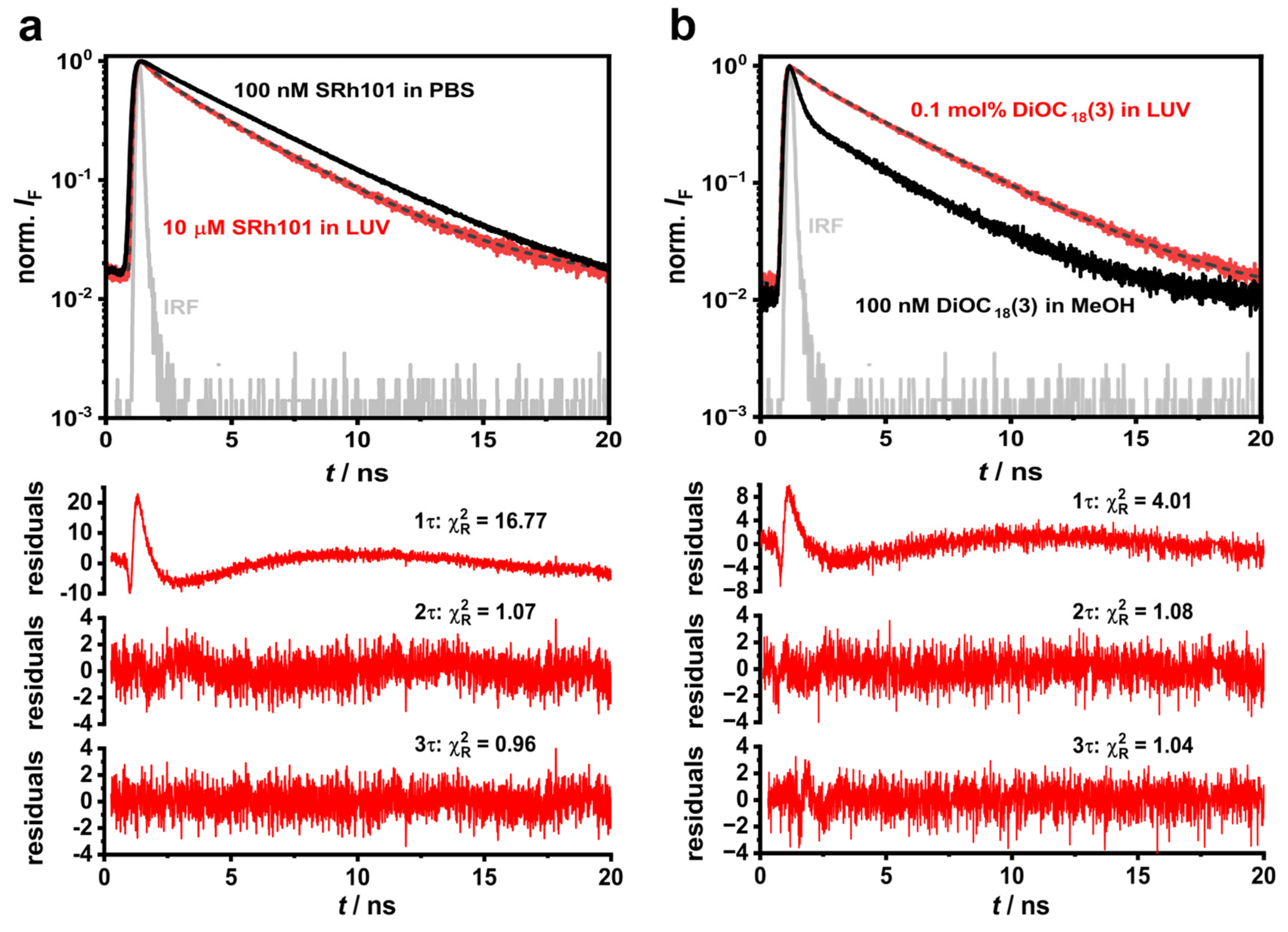
3.2. Time-Resolved Fluorescence Recordings of Fluorescently Labelled LUVs
3.3. Diffusion Characteristics of Fluorescently Labelled LUVs
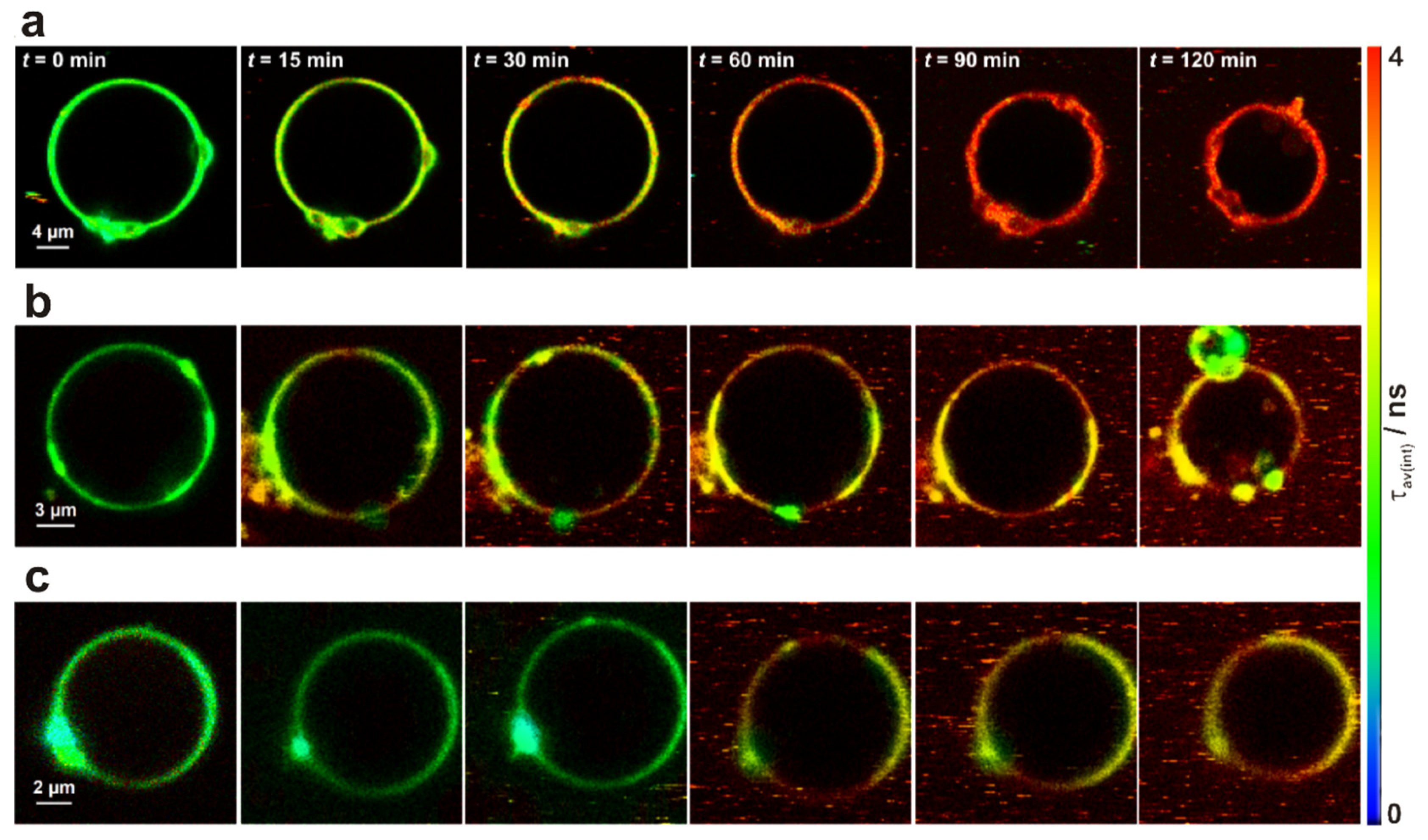
3.4. Interaction of Fluorescently Labelled LUVs and GUVs of Varying Lipid Composition Using 2P-FLIM Recordings
3.5. Interaction of Fluorescently Labelled LUVs and GUVs of Varying Lipid Composition Using Time-Resolved Fluorescence and 2P-FCS Recordings
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Farokhzad, O.C.; Langer, R. Impact of Nanotechnology on Drug Delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.; Templeton, A.K.; Munshi, A.; Ramesh, R. Nanodrug Delivery Systems: A Promising Technology for Detection, Diagnosis, and Treatment of Cancer. AAPS PharmSciTech 2014, 15, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Jahangirian, H.; Lemraski, G.E.; Webster, T.J.; Rafiee-Moghaddam, R.; Abdollahi, Y. A Review of Drug Delivery Systems Based on Nanotechnology and Green Chemistry: Green Nanomedicine. Int. J. Nanomed. 2017, 12, 2957–2978. [Google Scholar] [CrossRef]
- Hart, S.L. Lipid Carriers for Gene Therapy. Curr. Drug Deliv. 2005, 2, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Advanced Drug Delivery 2020 and beyond: Perspectives on the Future. Adv. Drug Deliv. Rev. 2020, 158, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Poste, G.; Papahadjopoulos, D.; Vail, W.J. Lipid Vesicles as Carriers for Introducing Biologically Active Materials into Cells. In Methods in Cell Biology; Prescott, D.M., Ed.; Academic Press: New York, NY, USA, 1976. [Google Scholar]
- Kozubek, A.; Gubernator, J.; Przeworska, E.; Stasiuk, M. Liposomal Drug Delivery, a Novel Approach: PLARosomes. Acta Biochim. Pol. 2000, 47, 639–664. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A Review on Phospholipids and Their Main Applications in Drug Delivery Systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Mentkowski, K.I.; Snitzer, J.D.; Rusnak, S.; Lang, J.K. Therapeutic Potential of Engineered Extracellular Vesicles. AAPS J. 2018, 20, 50. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Dagur, R.S.; Liao, K.; Peeples, E.S.; Hu, G.; Periyasamy, P.; Buch, S. Strategies for the Use of Extracellular Vesicles for the Delivery of Therapeutics. J. Neuroimmune Pharmacol. 2020, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, Y.; Wang, S.; Liu, J.; Hao, H. Extracellular Vesicles: Powerful Candidates in Nano-Drug Delivery Systems. Drug Deliv. Transl. Res. 2023, 14, 295–311. [Google Scholar] [CrossRef]
- Nagayasu, A.; Uchiyama, K.; Kiwada, H. The Size of Liposomes: A Factor Which Affects Their Targeting Efficiency to Tumors and Therapeutic Activity of Liposomal Antitumor Drugs. Adv. Drug Deliv. Rev. 1999, 40, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Dimova, R.; Aranda, S.; Bezlyepkina, N.; Vesselin, N.; Riske, K.A.; Lipowsky, R. A Practical Guide to Giant Vesicles. Probing the Membrane Nanoregime via Optical Microscopy. J. Phys. Condens. Matter 2006, 18, 1151–1176. [Google Scholar] [CrossRef] [PubMed]
- Dimova, R. Giant Vesicles and Their Use in Assays for Assessing Membrane Phase State, Curvature, Mechanics, and Electrical Properties. Annu. Rev. Biophys. 2019, 48, 93–119. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.J.; Smith, I.; Parker, I.; Bootman, M.D. Fluorescence Microscopy. Cold Spring Harb. Protoc. 2014, 2014, 1042–1065. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.A.; Shroff, H. Fluorescence Microscopy: A Concise Guide to Current Imaging Methods. Curr. Protoc. Neurosci. 2017, 79, 2.1.1–2.1.25. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Fluorescence Lifetime Imaging—Techniques and Applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence Lifetime Imaging Microscopy: Fundamentals and Advances in Instrumentation, Analysis, and Applications. J. Biomed. Opt. 2020, 25, 071203. [Google Scholar] [CrossRef] [PubMed]
- Schwille, P.; Haupts, U.; Maiti, S.; Webb, W.W. Molecular Dynamics in Living Cells Observed by Fluorescence Correlation Spectroscopy with One- and Two-Photon Excitation. Biophys. J. 1999, 77, 2251–2265. [Google Scholar] [CrossRef] [PubMed]
- Mütze, J.; Ohrt, T.; Schwille, P. Fluorescence Correlation Spectroscopy in Vivo. Laser Photonics Rev. 2011, 5, 52–67. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Hope, M.J.; Bally, M.B.; Webb, G.; Cullis, P.R. Production of Large Unilamellar Vesicles by a Rapid Extrusion Procedure. Characterization of Size Distribution, Trapped Volume and Ability to Maintain a Membrane Potential. Biochim. Biophys. Acta BBA—Biomembr. 1985, 812, 55–65. [Google Scholar] [CrossRef]
- Angelova, M.I.; Dimitrov, D.S. Liposome Electroformation. Faraday Discuss. Chem. Soc. 1986, 81, 303–311. [Google Scholar] [CrossRef]
- Makarov, N.S.; Drobizhev, M.; Rebane, A. Two-Photon Absorption Standards in the 550–1600 Nm Excitation Wavelength Range. Opt. Express 2008, 16, 4029. [Google Scholar] [CrossRef]
- Rumi, M.; Ehrlich, J.E.; Heikal, A.A.; Perry, J.W.; Barlow, S.; Hu, Z.; McCord-Maughon, D.; Parker, T.C.; Röckel, H.; Thayumanavan, S.; et al. Structure-Property Relationships for Two-Photon Absorbing Chromophores: Bis-Donor Diphenylpolyene and Bis(Styryl)Benzene Derivatives. J. Am. Chem. Soc. 2000, 122, 9500–9510. [Google Scholar] [CrossRef]
- Magde, D.; Wong, R.; Seybold, P.G. Fluorescence Quantum Yields and Their Relation to Lifetimes of Rhodamine 6G and Fluorescein in Nine Solvents: Improved Absolute Standards for Quantum Yields. Photochem. Photobiol. 2002, 75, 327–334. [Google Scholar] [CrossRef]
- Sjöback, R.; Nygren, J.; Kubista, M. Absorption and Fluorescence Properties of Fluorescein. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1995, 51, L7–L21. [Google Scholar] [CrossRef]
- Culbertson, C.T.; Jacobson, S.C.; Ramsey, J.M. Diffusion Coefficient Measurements in Microfluidic Devices. Talanta 2002, 56, 365–373. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy Principles of Fluorescence Spectroscopy, 3rd ed.; Springer Science & Business Media: New York, NY, USA, 2006. [Google Scholar]
- Kim, S.A.; Heinze, K.G.; Bacia, K.; Waxham, M.N.; Schwille, P. Two-Photon Cross-Correlation Analysis of Intracellular Reactions with Variable Stoichiometry. Biophys. J. 2005, 88, 4319–4336. [Google Scholar] [CrossRef]
- Haustein, E.; Schwille, P. Fluorescence Correlation Spectroscopy: Novel Variations of an Established Technique. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 151–169. [Google Scholar] [CrossRef]
- Heinze, K.G.; Koltermann, A.; Schwille, P. Simultaneous Two-Photon Excitation of Distinct Labels for Dual-Color Fluorescence Crosscorrelaion Analysis. Proc. Natl. Acad. Sci. USA 2000, 97, 10377–10382. [Google Scholar] [CrossRef]
- Bhowmik, B.B.; Basu, S.; Ray, D. Photophysical Studies of 3,3′ Dioctadecyloxacarbocyanine Dye in Model Biological Membranes and Different Solvents. Chem. Phys. Lipids 2001, 109, 175–183. [Google Scholar] [CrossRef]
- VanderMeulen, D.L.; Misra, P.; Michael, J.; Spears, K.G.; Khoka, M. Laser Mediated Release of Dye from Liposomes. Photochem. Photobiol. 1992, 56, 325–332. [Google Scholar]
- Albota, M.A.; Xu, C.; Webb, W.W. Two-Photon Fluorescence Excitation Cross Sections of Biomolecular Probes from 690 to 960 Nm. Appl. Opt. 1998, 37, 7352–7356. [Google Scholar] [CrossRef]
- Mütze, J.; Iyer, V.; Macklin, J.J.; Colonell, J.; Karsh, B.; Petrášek, Z.; Schwille, P.; Looger, L.L.; Lavis, L.D.; Harris, T.D. Excitation Spectra and Brightness Optimization of Two-Photon Excited Probes. Biophys. J. 2012, 102, 934–944. [Google Scholar] [CrossRef]
- Suhling, K.; French, P.M.; Phillips, D. Time-Resolved Fluorescence Microscopy. Photochem. Photobiol. Sci. 2005, 4, 13–22. [Google Scholar] [CrossRef]
- Chen, R.F.; Knutson, J.R. Mechanism of Fluorescence Concentration Quenching of Carboxyfluorescein in Liposomes: Energy Transfer to Nonfluorescent Dimers. Anal. Biochem. 1988, 172, 61–77. [Google Scholar] [CrossRef]
- Packard, B.S.; Wolf, D.E. Fluorescence Lifetimes of Carbocyanine Lipid Analogs in Phospholipid Bilayers. Biochemistry 1985, 24, 5176–5181. [Google Scholar] [CrossRef]
- Sims, P.J.; Waggoner, A.S.; Wang, C.-H.; Hoffman, J.F. Studies on the Mechanism by Which Cyanine Dyes Measure Membrane Potential in Red Blood Cells and Phosphatidylcholine Vesicles. Biochemistry 1974, 13, 3315–3330. [Google Scholar] [CrossRef]
- DeToma, R.P.; Brandt, L. Excited State Solvation Dynamics of 2-Anilinonaphthalene. Chem. Phys. Lett. 1976, 47, 231–236. [Google Scholar] [CrossRef]
- Matayoshi, E.D.; Kleinfeld, A.M. Emission Wavelength-Dependent Decay of the 9-Anthroyloxy-Fatty Acid Membrane Probes. Biophys. J. 1981, 35, 215–235. [Google Scholar] [CrossRef]
- Pramanik, A.; Thyberg, P.; Rigler, R. Molecular Interactions of Peptides with Phospholipid Vesicle Membranes as Studied by Fluorescence Correlation Spectroscopy. Chem. Phys. Lipids 2000, 104, 35–47. [Google Scholar] [CrossRef]
- Rigler, P.; Meier, W. Encapsulation of Fluorescent Molecules by Functionalized Polymeric Nanocontainers: Investigation by Confocal Fluorescence Imaging and Fluorescence Correlation Spectroscopy. J. Am. Chem. Soc. 2006, 128, 367–373. [Google Scholar] [CrossRef]
- Brewer, J.; Bloksgaard, M.; Kubiak, J.; Sorensen, J.A.; Bagatolli, L.A. Spatially Resolved Two-Color Diffusion Measurements in Human Skin Applied to Transdermal Liposome Penetration. J. Investig. Dermatol. 2013, 133, 1260–1268. [Google Scholar] [CrossRef]
- Pantazatos, D.P.; MacDonald, R.C. Directly Observed Membrane Fusion between Oppositely Charged Phospholipid Bilayers. J. Membr. Biol. 1999, 170, 27–38. [Google Scholar] [CrossRef]
- Lira, R.B.; Robinson, T.; Dimova, R.; Riske, K.A. Highly Efficient Protein-Free Membrane Fusion: A Giant Vesicle Study. Biophys. J. 2019, 116, 79–91. [Google Scholar] [CrossRef]
- Agudo-Canalejo, J.; Lipowsky, R. Critical Particle Sizes for the Engulfment of Nanoparticles by Membranes and Vesicles with Bilayer Asymmetry. ACS Nano 2015, 9, 3704–3720. [Google Scholar] [CrossRef]
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An Inverted Hexagonal Phase of Cationic Liposome-DNA Complexes Related to DNA Release and Delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef]
- Mora, N.L.; Boyle, A.L.; van Kolck, B.J.; Rossen, A.; Pokorná, Š.; Koukalová, A.; Šachl, R.; Martin, H.; Kros, A. Controlled Peptide-Mediated Vesicle Fusion Assessed by Simultaneous Dual-Colour Time-Lapsed Fluorescence Microscopy. Sci. Rep. 2020, 10, 3087. [Google Scholar] [CrossRef]
- Berezhna, S.; Schaefer, S.; Heintzmann, R.; Jahnz, M.; Boese, G.; Deniz, A.; Schwille, P. New Effects in Polynucleotide Release from Cationic Lipid Carriers Revealed by Confocal Imaging, Fluorescence Cross-Correlation Spectroscopy and Single Particle Tracking. Biochim. Biophys. Acta—Biomembr. 2005, 1669, 193–207. [Google Scholar] [CrossRef]
- Cavalcanti, R.R.M.; Lira, R.B.; Riske, K.A. Membrane Fusion Biophysical Analysis of Fusogenic Liposomes. Langmuir 2022, 38, 10430–10441. [Google Scholar] [CrossRef]
- Hope, M.J.; Mui, B.; Ansell, S. Cationic Lipids, Phosphatidylethanolamine and the Intracellular Delivery of Polymeric, Nucleic Acid-Based Drugs (Review). Mol. Membr. Biol. 1998, 15, 1–14. [Google Scholar] [CrossRef]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the Mechanism Whereby Cataionic Lipids Promote Intracellular Delivery of Polynucleic Acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef]
- Kim, B.-K.; Hwang, G.-B.; Seu, Y.-B.; Choi, J.-S.; Jin, K.S.; Doh, K.-O. DOTAP/DOPE Ratio and Cell Type Determine Transfection Efficiency with DOTAP-Liposomes. Biochim. Biophys. Acta—Biomembr. 2015, 1848, 1996–2001. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S.K. In Vitro Release Kinetics Model Fitting of Liposomes: An Insight. Chem. Phys. Lipids 2016, 201, 28–40. [Google Scholar] [CrossRef]
- Lin, A.J.; Slack, N.L.; Ahmad, A.; George, C.X.; Samuel, C.E.; Safinya, C.R. Three-Dimensional Imaging of Lipid Gene-Carriers: Membrane Charge Density Controls Universal Transfection Behavior in Lamellar Cationic Liposome-DNA Complexes. Biophys. J. 2003, 84, 3307–3316. [Google Scholar] [CrossRef]
- Bower, A.J.; Li, J.; Chaney, E.J.; Marjanovic, M.; Spillman, D.R.; Boppart, S.A. High-Speed Imaging of Transient Metabolic Dynamics Using Two-Photon Fluorescence Lifetime Imaging Microscopy. Optica 2018, 5, 1290–1296. [Google Scholar] [CrossRef]
- Karpf, S.; Riche, C.T.; Di Carlo, D.; Goel, A.; Zeiger, W.A.; Suresh, A.; Portera-Cailliau, C.; Jalali, B. Spectro-Temporal Encoded Multiphoton Microscopy and Fluorescence Lifetime Imaging at Kilohertz Frame-Rates. Nat. Commun. 2020, 11, 2062. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, Y.; Best-Popescu, C.; Gao, L. High-Speed Compressed-Sensing Fluorescence Lifetime Imaging Microscopy of Live Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2004176118. [Google Scholar] [CrossRef]
- Hanley, Q.S.; Arndt-Jovin, D.J.; Jovin, T.M. Spectrally Resolved Fluorescence Lifetime Imaging Microscopy. Appl. Spectrosc. 2002, 56, 155–166. [Google Scholar] [CrossRef]
- Niehörster, T.; Löschberger, A.; Gregor, I.; Krämer, B.; Rahn, H.-J.; Patting, M.; Koberling, F.; Enderlein, J.; Sauer, M. Multi-Target Spectrally Resolved Fluorescence Lifetime Imaging Microscopy. Nat. Methods 2016, 13, 257–262. [Google Scholar] [CrossRef]
- Park, J.; Gao, L. Advancements in Fluorescence Lifetime Imaging Microscopy Instrumentation: Towards High Speed and 3D. Curr. Opin. Solid State Mater. Sci. 2024, 30, 101147. [Google Scholar] [CrossRef]
- Stater, E.P.; Sonay, A.Y.; Hart, C.; Grimm, J. The Ancillary Effects of Nanoparticles and Their Implications for Nanomedicine. Nat. Nanotechnol. 2021, 16, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Najer, A.; Rifaie-Graham, O.; Yeow, J.; Adrianus, C.; Chami, M.; Stevens, M.M. Differences in Human Plasma Protein Interactions between Various Polymersomes and Stealth Liposomes as Observed by Fluorescence Correlation Spectroscopy. Macromol. Biosci. 2023, 23, e2200424. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruun, K.; Löhmannsröben, H.-G.; Hille, C. Benefits of Combined Fluorescence Lifetime Imaging Microscopy and Fluorescence Correlation Spectroscopy for Biomedical Studies Demonstrated by Using a Liposome Model System. Biophysica 2024, 4, 207-226. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica4020015
Bruun K, Löhmannsröben H-G, Hille C. Benefits of Combined Fluorescence Lifetime Imaging Microscopy and Fluorescence Correlation Spectroscopy for Biomedical Studies Demonstrated by Using a Liposome Model System. Biophysica. 2024; 4(2):207-226. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica4020015
Chicago/Turabian StyleBruun, Kristina, Hans-Gerd Löhmannsröben, and Carsten Hille. 2024. "Benefits of Combined Fluorescence Lifetime Imaging Microscopy and Fluorescence Correlation Spectroscopy for Biomedical Studies Demonstrated by Using a Liposome Model System" Biophysica 4, no. 2: 207-226. https://0-doi-org.brum.beds.ac.uk/10.3390/biophysica4020015