1. Introduction
In biocatalysis, numerous reactions cannot be completed by a single enzyme and often require two or more enzymes to achieve cascade reactions. A variety of functionally related enzyme molecules are precisely assembled to establish multi-enzyme complexes (MECs), such that the enzyme molecules are close to each other in space, to form substrate channels, which can enhance the synergy and catalytic efficiency of cascade enzymes [
1].
In recent years, with advancements in synthetic biology research and development, researchers have used microbial cells or cell-free systems to program and reorganize multi-enzyme systems in metabolic pathways and have synthesized numerous functional compounds, reducing metabolic efficiency and output. Various natural multi-enzyme self-assembly complexes exist in organisms, such as the cellulosome machinery and kinase cascade pathways involved in cell signal transduction [
2,
3,
4]. The substrate channel effect and synergistic mechanism in such systems are responsible for the high catalytic efficiency of multi-enzyme complexes. Simulating and borrowing from natural multi-enzyme systems and combining the interactions of proteins with DNA and RNA in organisms to design and construct artificial self-assembled multi-enzyme systems are key strategies of improving metabolic efficiency [
5,
6].
Multi-enzyme complexes are products of natural evolution where several different enzymes form a single unit in terms of structure and function. This can shorten the distance between multiple enzymes and improve the transfer efficiency of intermediate reaction products between adjacent cascade enzymes (substrate shuttle effect), thereby improving the overall reaction efficiency and promoting cascade biocatalysis. Inspired by nature, researchers have developed various methods of assembling artificial multi-enzyme complexes with different spatial structures to improve the catalytic efficiency of biological cascades [
7]. For example, a combined cross-linked enzyme aggregate (combi-CLEA) is constructed using a chemical cross-linking method, and multiple enzymes are expressed in the form of a fusion enzyme by constructing a fusion gene. Alternatively, a multi-enzyme complex can be constructed by fusing multiple enzymes with affinity domains (such as the PDZ domain and its ligands, the leucine zipper domain, and SpyTag/Catcher) [
8,
9,
10,
11] using the affinity between domains. Based on the advantages of strong operability, high product yield, and fast reaction speed of the in vitro multi-enzyme system, starch and inorganic ammonia have been converted into amino sugars through phosphorolysis-isomerization-amination-dephosphorylation of the five core enzymes. Owing to the thermodynamically prone irreversible dephosphorization reaction catalyzed by the dephosphorization enzyme (GlmP), a multi-enzyme system can theoretically achieve the maximum conversion from starch to amino sugars. Through a database search and literature analysis, we excavated and obtained enzyme elements with high enzyme activity and expression levels and consistent reaction conditions. The currently reported dephosphorylase GlmP, with the highest substrate specificity for GlcN6P (GlcN6P), was obtained through careful screening of the key dephosphorylase enzymes [
12]. In recent years, scaffold-based artificial multi-enzyme complexes with controllable spatial structures have been gradually constructed [
13,
14].
Certain anaerobic microorganisms in nature can produce a multi-enzyme complex for organizing and coordinating multiple enzyme components to synergize and efficiently catalyze cellulose degradation [
15]. Such complexes are called cellulosomes and are considered miniature and efficient cellulose degradation machines [
16]. At present, although an overall understanding of the complex system is still lacking, the assembly of cellulase into cellulosomes is considered the most effective strategy of degrading cellulose in microbial systems, while also being an important method of developing and utilizing cellulose [
17]. Cellulosomes were first isolated and identified from
Clostridium thermocellum by Lamed et al. [
18] and subsequently discovered in and isolated from anaerobic bacteria and fungi [
19]. As these anaerobic microorganisms are not easy to cultivate and the molecular weight of natural cellulosomes is relatively large (650–2500 ku) [
20], the separation and purification method remains cumbersome [
21], which greatly limits their effective application. Following continuous extensive research on the structure and function of cellulosomes, the concept of artificially designed cellulosomes has been proposed in recent years [
22,
23]. To achieve the artificial design of cellulosomes, DNA recombination technology is used to clone, fuse, and express the scaffold protein and cellulase genes carrying the anchor domain. After the recombinant protein is purified, the cellulase carrying the corresponding anchor domain is reconstructed into a cellulosome by the scaffold protein in vitro [
24,
25,
26]. However, the production cost is a limiting factor when only purified recombinant proteins are used for in vitro remodeling of cellulosomes [
27,
28]. Genetic engineering technologies can be used to design cellulosomes with specific functions and to induce high secretion and expression of each component protein in the host bacteria. The expression product self-assembles into artificial cellulosomes outside the cell to improve the degradation of complex polysaccharides. This is a relatively inexpensive and convenient method of cellulose degradation [
29].
The key non-catalytic subunit in cellulosomes is scaffoldin, which contains multiple cohesins that interact specifically with the docking module (dockerin) of the enzyme. Scaffoldin stabilizes various enzymes in the supramolecular structure of the complex. Studies have found that the efficient self-assembly of cellulosomes outside the cell requires the participation of a certain concentration (0.5–2 mM) of Ca
2+ [
30,
31]. However, owing to metabolic requirements, the concentration of Ca
2+ in microbial cells at rest is generally 10
−4 mM, which is much lower than the extracellular concentration (1 mM). This is strictly controlled by Ca
2+ channels and other components that limit the self-assembly process of cellulosomes and their cellular components. As previous research has shown that it is difficult to achieve a high-efficiency assembly of intracellular multi-enzyme systems, the aim of this study was to learn from the model of efficient assembly of extracellular cellulosomes, optimize the design of key components to reduce the environmental dependence on Ca
2+, and achieve a more efficient intracellular multi-enzyme assembly process for the efficient production of amino acids.
The present study selected cellulosomes as the basic model of the intracellular self-assembly system and its key components (docking protein DocA and adhesion protein Coh) as the main research object. Using Escherichia coli as a platform, we focused on exploring and analyzing its self-assembly mechanism. Here, we focused on the Ca2+-binding region of the docking protein DocA. Based on semi-rational design and high-throughput screening technology, we designed a docking protein that does not rely on Ca2+ and obtained three site-directed mutant proteins: DocA-S1, DocA-S2, and DocA-S3. Molecular dynamics simulation analysis was performed, and combined experiments were conducted to determine the mutation direction suitable for intracellular assembly. To address the key points of the cellulosomes, docking protein mutants that did not rely on Ca2+ were obtained gradually and assembled with Coh at low intracellular Ca2+ concentrations to achieve the self-assembly of cellulosome key components without relying on Ca2+.
2. Materials and Methods
2.1. Strains, Vectors, Chemicals, Media, and Culture Conditions
E. coli BL21 (DE3) was used as an expression host and cultured in Luria Broth (LB) medium at 37 °C. The pET28a(+) and pETDuet-1 vectors (Sangon, Shanghai, China) were used for gene cloning. Enzymes used for DNA amplification and restriction were purchased from Vazyme (Nanjing, China). Primers were synthesized by Qingke (Beijing, China). Part of the fragment synthesis applied to BIFC-FC was synthesized using GenScript. The plasmid preparation kit was purchased from Vazyme. All the chemicals were purchased from Sigma-Aldrich. The strains and plasmids used are listed in
Table 1. The construction process and schematic diagram of the recombinant plasmid are presented in
Figures S1–S3.
2.2. Selection of the Key Components of the Cellulosome
In the present study, the natural multi-enzyme complex cellulosome was selected as a research model for an intracellular self-assembly system, primarily focusing on the intracellular assembly of docking and adhesion proteins. Using the NCBI (
https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 16 March 2023) and PDB (
http://www.rcsb.org, accessed on 16 March 2023) databases, we downloaded the amino acid sequences and crystal structures, respectively. PyMOL analysis was used to select docking proteins (DocA, PDB ID: 2CCL) and adhesion proteins (Coh, PDB ID code: 1OHZ) with existing crystal structures. In the original assembly of the intracellular self-assembly system, both proteins were derived from
C. thermocellum.
2.3. Mutation Site Selection of Docking Protein DocA
A combination of molecular simulation dynamics and molecular biology technology was used to mutate the DocA protein, selected due to its small structure and high operability. The amino acids within 4 Å of the Ca
2+ in the Ca
2+ binding site of the protein DocA were used as the key amino acids for Ca
2+ binding, including D5, N7, D9, D16, D39, D41, N43, and D50. To minimize the mutation screening workload, ISM evolution design software CASTER 2.1 was used to analyze the composition of pseudo-mutant amino acids. Finally, NYS was selected as a degenerate codon for the saturation mutation (including Thr and Ser, whose side chains were similarly hydrophilic but uncharged, and are hydrophobic amino acids that facilitate folding). GROMACS, PyMOL, and CASTER 2.1 were used to design and simulate the key amino acids. Sixteen docking proteins with single point mutations were obtained; the sequences of each protein are shown in
Table 2.
2.4. Molecular Dynamics (MD) Simulations
MD simulations were performed using the GROMACS 4.5.4 suite (
http://www.gromacs.org/, accessed on 20 May 2023) with a GROMOS 96 force field and SPC/E explicit water system. The net charge of the system was neutralized using sodium ions. The protein is placed in a square box, the edge of the box is 1.5 nm away from the protein, and 15,000 water molecules are added to solvate the protein. The system was minimized in two stages with constrained and unconstrained optimizations: an initial minimization with the steepest descent (maximum force of 50 kJ/mol/nm in 1500 steps), followed by minimization with the conjugate gradient algorithm (maximum force of 10 kJ/mol/nm in 2000 steps) [
32].
An integration step of 0.002 ps was used and the bonds were constrained using the LINCS algorithm [
33]. The minimized system was gradually equilibrated at 300 K for 100 ps with restrained proteins and ligands. Subsequently, a 10-ns equilibration stage was adopted without restraint.
2.5. Mutation Design and Verification of Docking Protein
After the GROMACS fitting operation, the mutant with the smallest RMSD was selected for experimental verification. The protein sequences are listed in
Table 3. Based on the calculations, the amino acid sequence was optimized according to the
E. coli expression system. Mutation primers for Ca
2+ binding to key amino acids were designed (forward and reverse primers, respectively, covering the DocA Ca
2+ binding site) and site-directed mutagenesis technology was used to perform mutations to obtain docking protein DocA mutants. The DocA mutants were obtained using a rapid site-directed mutagenesis kit. The specific mutation primer sequences are listed in
Table 4.
2.6. Expression and Purification of DocA and Coh
The verified strain was cultured and activated overnight in 50 mL liquid LB medium containing kanamycin (50 μg × mL−1). Then, 1% of the inoculum was inoculated into liquid LB medium. When the culture reached an OD600 ≈ 1.0, IPTG was added to a final concentration of 0.2 mM. Bacterial solution was induced at 26 °C for 10 h. The induced bacterial cells were collected and then 400 mL of bacterial liquid centrifugal enrichment was resuspended in 10 mL of 2× PBS buffer. An ultrasonic disintegrator was used to disrupt the cells. Because later experiments had certain requirements for protein purity, the His Tag Protein Purification Kit (Beyotime, Haimen, China) was used for purification. The entire operation was carried out in a 4 °C chromatography cabinet.
SDS-PAGE was used to detect the protein purity of the purified proteins. The proteins that met the requirements were collected together. DocA and Coh were desalted using a concentrated treatment of PBS-EP + buffer dialysis salt for protein DocA, and acetic acid-acetate buffers of different pH + buffer dialysis salt for Coh.
2.7. Detection of the Binding Affinity of DocA and Coh Proteins
The Biacore T200 molecular interaction analyzer (Malborough, MA, USA) was used to explore the binding mechanism of the two proteins. Coh protein was anchored on a suitable chip, and DocA was mixed with different concentrations of CaCl2 to flow through the chip.
According to the operation manual, an appropriate M5 chip was selected as the anchor chip per the manufacturer’s instructions, and the approximate required concentration of protein Coh was calculated. The gradient was diluted using acetate-acetate buffers of different pH similar to the anchoring protein. The optimal anchor concentration and pH were determined according to the anchor case, and the protein was aligned according to the “capture method” in the “Biacore Small Molecular Application Manual”.
The anchored chip was loaded into the molecular interaction, and the buffer was made up of 1× PBS solution. DocA protein was diluted to the Coh (anchor protein) concentration until the concentration was almost uniform, and different concentrations of CaCl2 were combined. The sample was placed at 4 °C for at least 30 min. Binding affinity was measured using Biacore T200, and the binding status was evaluated based on the AbsResp value. In order to ensure the accuracy of the data, three biological repeats were carried out in this part of the experiment, and the experimental samples were set up in parallel three times.
2.8. Application of Bimolecular Fluorescence Complementation-Flow Cytometric Sorting (BIFC-FC)
BIFC-FC was used to verify the interaction effects of different docking protein mutants and cohesin Coh in vivo. The yellow fluorescent protein (eYFP) was selected and divided into two fragments, eYFP-N (1–155) and eYFP-C (156–238), and connected to the docking protein DocA and cohesin Coh through the longer connecting peptide SGGGSGGGSGGS. The N-terminal of Coh and C-terminal of DocA were selected to participate in the fusion of eYFP so that the fragment was more stable. After ligation, it was simultaneously expressed on the plasmid pETDuet-1, and transformation was verified. If there was an interaction between the docking protein mutant and cohesin Coh, the N-terminal and C-terminal fragments of the fluorescent protein can be drawn close to each other through the linker, and the fluorescent protein chromophore is formed for re-fluorescence. Fluorescence signals were screened using the MoFlo XDP ultra-speed flow cytometry sorting system (Beckman Coulter Co., Ltd., Indianapolis., IN, USA). The construction process and a schematic diagram of the recombinant plasmid are shown in
Figures S4–S6.
2.9. Assembly and Detection of Key Enzymes in L-Lysine Fermentation
For L-lysine-producing bacteria E. coli QDE, pyruvate was accumulated by glycolysis with glucose as substrate. After pyruvate was converted to oxaloacetic acid, the effect of aspartate aminotransferase (encoded by the aspC gene) was enhanced to maximize the anabolic flow to L-aspartic acid. The key enzymes involved are, in order: Aspartic kinase AK (lysC gene encoding), Aspartic semi-aldehyde dehydrogenase ASADH (asd gene encoding), dihydropyridine dicarboxylate synthase DHDPS (dapA gene encoding), dihydropyridine dicarboxylate reductase DHDPR (dapB gene encoding), n-succinyl-L diaminoheptadecarboxylate desuccinylase (dapE gene encoding), diaminopimaric acid differential isomerase (encoded by the gene dapF) and diaminopimaric acid decarboxylase DAPDC (encoded by the lysA gene).
The key components of cellulosomes, DocA-S3 and Coh, were connected to the key enzymes of L-lysine fermentation, aspartate semialdehyde dehydrogenase (Asd), dihydropyridine dicarboxylate synthetase (DapA), and dihydropyridine dicarboxylate reductase (DapB) in
E. coli through the linking peptide SGGGSGGGSGGS and were cloned into the pETDuet-1 plasmid for co-expression. The recombinant plasmid was then transferred to L-lysine-producing
E. coli QDE to obtain
E. coli QDE-DocA-S3 engineered bacteria. Fermentation conditions were as follows: 37 °C; pH 6.7; 300 r/min; glucose concentration = 2.0%; dissolved oxygen = 20–35%; and 25% ammonia water in a continuous flow. After induction, the fermentation time was less than 40 h, with sampling and testing every 4 h. The concentration of bacterial cells was determined using an ultraviolet spectrophotometer (UV-6100, METASH, Shanghai, China) to detect the absorbance of the sample at 600 nm after dilution. Glucose and L-lysine contents were measured using a biosensor (BSA-90, Jinan Yanke Co., Ltd., Jinan, China) [
34].
3. Results and Discussion
3.1. Mutation Strategy and Molecular Docking
Through molecular dynamics simulation analysis, Ca
2+ was observed to have the capacity to maintain the stability of the loop region in the key element of the cellulosome-docking protein, facilitating the interaction of the double α-helix connected to the loop region in the docking protein with the adhesion protein at a certain angle. Based on this, a semi-rational design method was used to directly modify the Ca
2+ binding site of the docking protein. Considering the hydrophilicity and hydrophobicity of the amino acids, 16 amino acid single point mutations were designed for D5, N7, D9, and D16. Swiss-model, GROMACS, PyMOL, and other molecular dynamics simulation software were used to carry out modeling, static, and dynamic combined analyses, and a schematic diagram of the process is presented in
Figure 1.
3.2. Simulation Calculation and Selection of Docking Protein DocA Mutation Site
GROMACS 4.5 was used to perform a 10-ns molecular dynamics simulation. To comprehensively analyze the degree of difference between each mutant protein and the original DocA, the g_rms and g_rmsf tools in GROMACS 4.5 software were used to analyze the structural difference parameters (RMSD value). The RMSD values for each calculation stage were collected and plotted on a scatter diagram (
Figure 2). The size and dispersion of data in
Figure 2 suggest that if the original DocA structure is combined with Coh alone in an environment without Ca
2+, the fluctuation is very obvious, the high frequency RMSD is about 0.38 nm, and the structural stability is very poor. If DocA-2Ca is combined with Coh in an environment containing Ca
2+, the high frequency RMSD drops to 0.26 nm and the structural stability improves significantly. According to the data size and dispersion, the RMSD of the mutant protein DocA-D5R was small and the structure was stable during the calculation process.
In the scatter plot of the RMSD, only the general data status can be observed. To further evaluate the structural similarity between the 16 mutant proteins and the original protein, the average RMSD values of the 16 docking protein mutants were calculated (
Table 5). On the one hand, in the calculation process, the original protein DocA lacked Ca
2+, the structure was extremely unstable, and the average RMSD was high, at 0.303947. The overall RMSD values of the mutant proteins DocA-D5Q, DocA-N7K, DocA-N7E, DocA-D9Q, DocA-D16K, and DocA-D16E were larger and higher than that of the original DocA. In other words, the structural difference from the original protein was the largest, the degree of dispersion was high, and the structure was unstable. In contrast, the original protein, DocA-2Ca, was theoretically stable, with an average RMSD of 0.270246. The mutant proteins DocA-D5R, DocA-D5K, DocA-D9R, DocA-D9E, and DocA-D16Q had lower RMSD values, which were lower than those of the original protein DocA-2Ca and tended to be more stable. Among them, the mean RMSD of the docking protein mutant DocA-D5R was the lowest (0.232006), and its structure had the highest structural similarity to the original protein DocA.
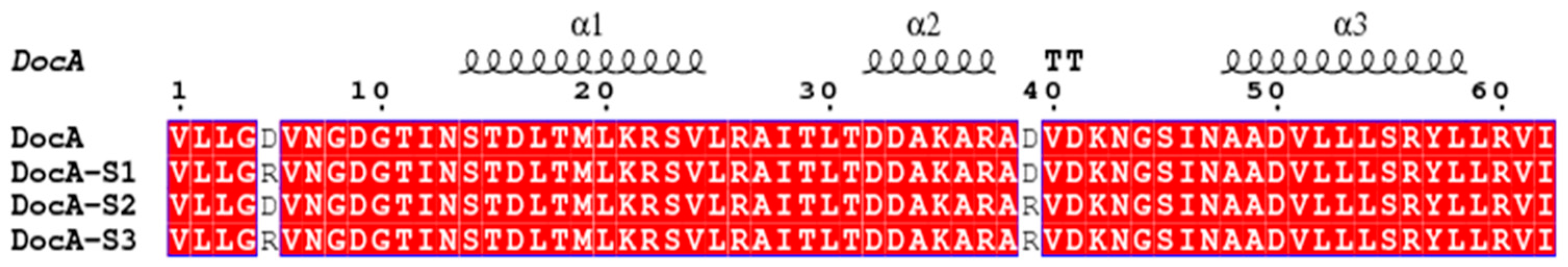
Corresponding to the docking protein mutant, DocA-D5R, DocA was located at the D36 position of the second binding site of DocA. We predicted that its mutation stability would be improved, and designed it to mutate D5 and D39 simultaneously. As shown in
Figure 3, the mutant proteins DocA-D5R, DocA-D39R, and the double-site combination mutants were named DocA-S1, DocA-S2, and DocA-S3, respectively. Three mutant proteins were obtained by site-directed mutagenesis of DocA. To verify the mutation effect, the changes in the binding ability of the docking protein mutants and cohesin Coh were determined in a low Ca
2+ environment and in vitro and in vivo experiments conducted.
3.3. In Vitro Verification of the Interaction between Mutant DocA and Protein Coh
According to the three-dimensional structures of the docking and adhesion proteins, there are two Ca2+-binding sites in the docking protein, and Ca2+ also plays an important role in the formation of a stable structure. Early experiments found that the Ca2+ concentration required for the interaction of docking protein DocA and cohesin Coh to form a stable structure was around 5 × 10−3 M; however, the intracellular Ca2+ concentration was much lower than the extracellular Ca2+ concentration. Therefore, we focused on exploring whether the mutation of DocA in a low-calcium environment can reduce Ca2+ dependence and enhance its binding ability to cohesin Coh.
The mutation sites of key amino acids for Ca
2+ binding of DocA were selected, and GROMACS 4.5 software was used for molecular dynamics simulation to preliminarily determine their influence on the structural stability of DocA. After site-directed mutagenesis technology was used, the successfully expressed docking protein, DocA, its mutants, and cohesin Coh were purified in vitro. After the purified protein was diluted, a Biacore T200 molecular interaction analyzer was used for computer testing. By collecting and analyzing the AbsResp value (i.e., the binding interaction value), we analyzed the binding ability of the docking protein mutant DocA with Coh, which is a Ca
2+ buffer solution with different concentration gradients, to determine the change in the degree of Ca
2+ dependence and analyze the mutation effect. The results are shown in
Figure 4.
Statistical analysis revealed the changes in the binding affinity of the docking protein and its mutants with the adhesion protein under different Ca2+ concentrations: (1) When the Ca2+ concentration was 10−7–10−4 M, the binding ability of the mutant proteins DocA-S1 and DocA-S3 to the cohesin Coh was improved greatly compared with that of the original DocA protein. The AbsResp values DocA-S1 < DocA-S3 were 2.1 and 4.3 times that of the binding capacity of the original DocA and Coh proteins, respectively, and exhibited a constantly increasing trend. The mutant DocA-S3 has the strongest binding ability with Coh, which was 3.8–4.6 times that of DocA. However, the binding affinity of the mutant protein DocA-S2 to the cohesin Coh was significantly weaker than that of the original DocA protein. (2) When the Ca2+ concentration was 10−4–10−3 M, the binding capacity of each protein to cohesin Coh was as follows: DocA-S3 > DocA-S1 > DocA > DocA-S2. The binding capacity of the mutant proteins DocA-S1 and DocA-S3 to the adhesion protein Coh reached a high level when the Ca2+ concentration was 10−4, and then, with an increase in the Ca2+ concentration, the AbsResp value changed slightly compared with the original protein. The ability of DocA to bind to Coh remained stable. The binding capacity of the mutant protein DocA-S2 and cohesin Coh was improved significantly but was still weaker than that of the original protein DocA. In summary, the binding ability of each docking protein to the adhesion protein Coh was initially enhanced and then stabilized gradually. The double-site mutant protein DocA-S3 performed best, and the binding affinity to the adhesion protein Coh was four times that of the original protein DocA. The single-point mutant protein DocA-S1 performed second-best, with a binding affinity to the adhesion protein Coh approximately twice that of the original protein DocA, and the single-point mutant protein DocA-S2 had a lower binding affinity than the original protein DocA. Therefore, the double-site mutant protein DocA-S3 was the best mutant.
3.4. In Vivo Verification of the Interaction between Mutant DocA and Coh Protein
Katsuaki et al. reported the in vitro assembly and cellulolytic activity of a β-glucosidase-coupled cellulosome complex, which contains three main cellulase enzymes and the thermal fiber of
Ruminiclostridium. The full-length scaffold protein and the dockerin module of
Thermocellum β-glucosidase-
C. thermocellum were fused. The results indicate that the enzymatically proportionally assembled β-glucosidase-integrated cellulosome complex is essential for the effective saccharification of crystalline cellulose [
35]. Notably, quantitative high-content imaging analysis showed that the direction of glucose flux between glycolysis, pentose phosphate pathway, and serine biosynthesis appears to be regulated spatially by the multi-enzyme complex in a cluster-size-dependent manner [
36]. The multi-step cascade reaction in nature, such as the multi-enzyme metabolic complex of glucose metabolism in living human cells, maximizes the efficiency of the reaction by assembling related enzymes together. This organization is conducive to the processing of intermediates by downstream enzymes. Previous studies on multi-enzyme nanocomposites assembled on DNA scaffolds have shown that closer distance between enzymes can improve the overall reaction efficiency [
37]. Our experimental concept was to shorten the process of transferring and processing intermediate metabolites between different enzyme catalytic active centers using multi-enzyme fusion, self-assembly, and other methods to construct multi-enzyme complexes in cells. Indirectly, this increased the catalytic rate of each enzyme, thereby increasing the synthesis efficiency of the product.
Bimolecular fluorescence complementation (BiFC) analysis technology uses the characteristics of the fluorescent protein and its mutants as reporter genes to divide the fluorescent protein into two molecular fragments with no fluorescence activity and to then connect the fragments to the target protein separately. If the two target proteins interact, the two molecular fragments of the fluorescent protein will also be brought close to each other spatially, and the active fluorescent group will be reformed to emit fluorescence. Bimolecular fluorescence complementation can be observed under a fluorescence microscope to determine whether two target proteins interact, the stability of the protein complex, and the influence of cell-signaling molecules on their interactions. This information is important for studying protein interactions.
Morell et al. [
38,
39,
40] used flow cytometry to detect the fluorescent signal of the BiFC fluorescent protein complex and achieved a real-time detection of protein-protein interaction in the cell. The method they described is BiFC-FC. As the flow cytometer (FACS) can quantitatively analyze the number of positive cells and the average fluorescence intensity of the fluorescence signal, and can detect a weaker fluorescence signal, its detection result is more reliable than that of the traditional fluorescence microscope observation method. At present, the application of flow cytometry technology to quantitatively analyze the BiFC signal in a single cell is becoming a standard method for living cell BiFC experiments. To verify the interaction effects of different docking protein mutants and cohesin Coh in vivo, we used eYFP for BIFC-FC and divided it into two fragments. The mutant DocA and the cohesive Coh protein were respectively connected by a longer connecting peptide, and the above fusion protein was expressed and verified by fermentation. At the same time, the fusion protein of SpyCatcher/SpyTag was designed for comparison and analysis of the interacting peptide. A flow cytometer was used to quantitatively analyze the number of positive cells and the average fluorescence intensity of the fluorescence signal; the results are shown in
Figure 5.
BIFC-FC was used to further analyze the assembly of mutants and Coh in cells. The number of positive cells and the average fluorescence intensity of the fluorescence signal were quantitatively analyzed using flow cytometry. As shown in
Figure 5 and
Figure 6, the fluorescence intensities of DocA-S1, DocA-S2, and DocA-S3 were 8.00 × 10
6, 2.30 × 10
6, and 1.50 × 10
7, respectively (among them, the fluorescence intensity of DocA was extremely weak and much lower than 10
5). The results showed that the binding affinities of DocA-S1, DocA-S2, and DocA-S3 to Coh in the cells were improved significantly. Considering that DocA mutants and Coh are bonded mainly through hydrogen bonds, their binding strength is second only to that of SpyCatcher/SpyTag, which is composed of chemical bonds. Doca-S3 had the maximum binding strength with Coh in a low intracellular calcium environment, which was significantly higher than that of DocA, indicating that it can significantly interact with Coh in the intracellular environment. This allows the key components of cellulosomes to be used beyond cell surface biomass degradation [
41] and in high-value chemical product synthesis [
42,
43,
44,
45,
46].
The intracellular fluorescence intensity of the three-point mutant proteins was significantly higher than that of the original proteins, and there was some overlap between the intensities of the interacting polypeptide pairs, in which the signal intensity of the mutant protein DocA-S3 was prominent. This indicates that the BIFC-FC-based screening scheme constructed in the present study can effectively screen for mutations at key sites involved in DocA Ca2+ binding.
3.5. Molecular Dynamics Simulation and Structural Analysis
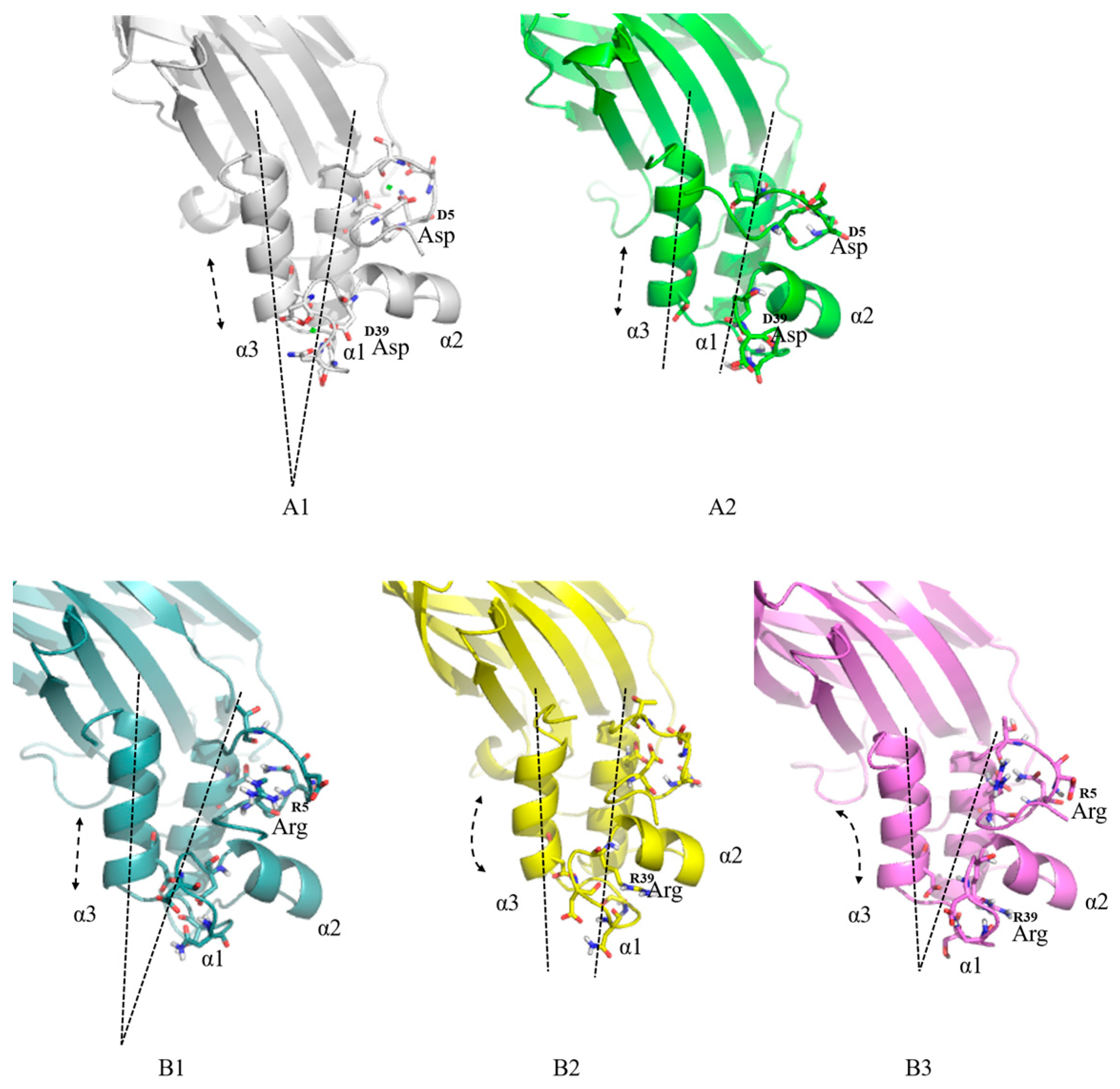
Different docking protein-cohesin (DocA, -CohA) complex structures were constructed for the above three docking protein mutants, DocA-S1, DocA-S2, and DocA-S3. Subsequently, to perform 10-ns molecular dynamics simulation, GROMACS 4.5 software was used and PyMol was used to compare the structure of the mutant and the original DocA protein, as shown in
Figure 7. The simulated structural changes in the mutant protein under the action of the GROMOS 96 force field are shown in
Figure 7. Detailed analysis and comparison of MD simulations showed that there were spatial gap changes between the mutant proteome and the pro-protein, mainly in the position shift of the ring. Through comparative analysis, as shown in
Figure 7A1,A2, the α1 and α3 helices of the original DocA existed stably at a fixed Angle when Ca
2+ is present. In the absence of Ca
2+, the α1 and α3 helices were loose and the displacement was obvious. Comparative analysis of the mutant proteins is shown in
Figure 7B1–B3. The α1 and α3 helices of the mutants DocA-S1 and DocA-S3 can form stable bonds with Coh (without Ca
2+) at an angle. Regarding the change in amino acid position, the Asp5 mutation of the initial DocA to Arg5 made the ring region of the calcium-binding site more compact, as observed in mutants DocA-S1 and DocA-S3. Among these, DocA-S3 combined two mutations, Asp5 and Asp39, making its interaction with Coh more stable.
The g_rms and g_rmsf tools in GROMACS 4.5 were used to analyze the difference parameters (RMSD values) between the mutant structure and the initial docking protein DocA structure, and the RMSD at each calculation stage was collected and scatter plots were constructed (
Figure 8). The results showed that out of all mutants, the RMSD data of DocA-S3 suggest this version was the most stable, including in structure. The mean RMSD values for the three docking protein mutants were calculated. The mean RMSD of the mutant DocA-S3 was the lowest at 0.2916, and its structure was the most stable, close to the stable state of the original protein DocA-2Ca at 0.29008. The mean RMSD value was 0.2955, followed by DocA-S1. Root-mean-square fluctuations (RMSF) were used to measure the mean amino acid flexibility of DocA mutants from the initial frame, as shown in
Figure 9. The fluctuation degree of DocA is large, and the RMSF value is high, which may also be the reason why the key amino acids of calcium ions are more prone to stable interaction with Coh after mutation. Molecular dynamics simulations and conformational analysis showed that in the absence of Ca
2+ addition, the D5R mutation in DocA-S3 could form a new hydrogen bond network with Gly and Val, ensuring that the double α-helix maintained the stability of its interaction with Coh at a certain angle and formed an intracellular self-assembly complex independent of Ca
2+.
3.6. Efficient Assembly and Detection of Key Enzymes for L-Lysine Production
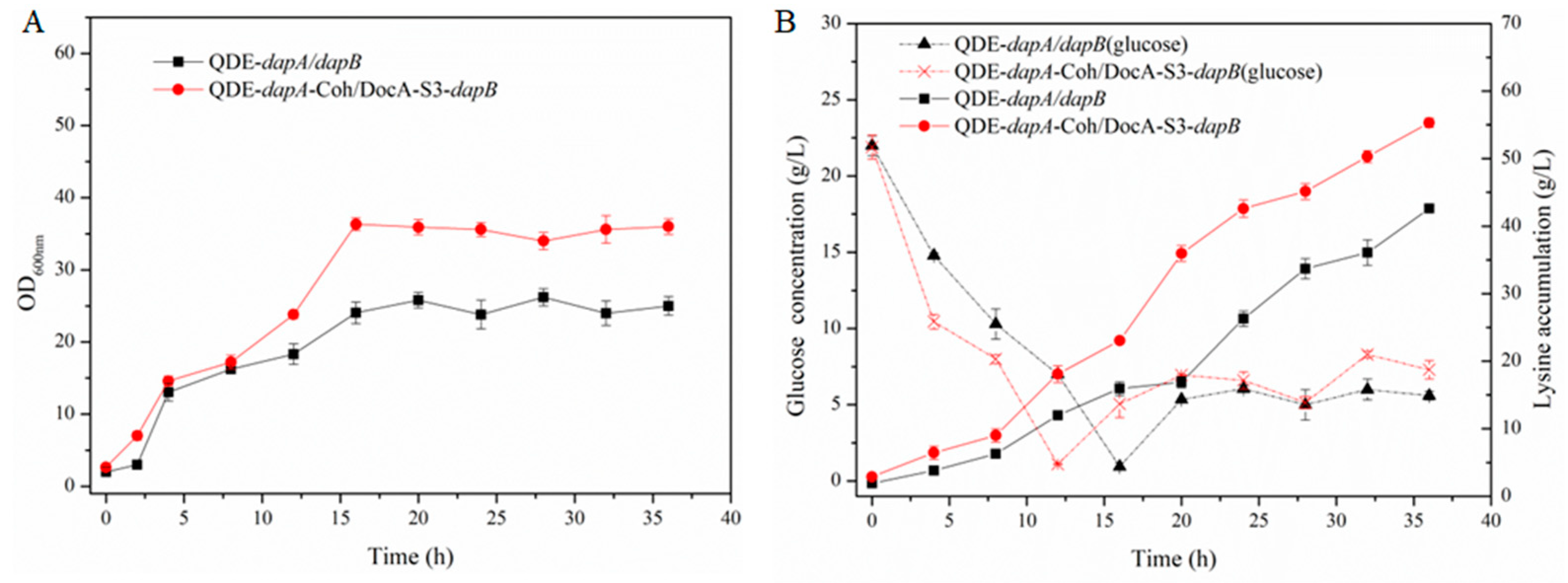
The key components of cellulosomes, DocA-S3 and Coh, were linked to the key enzymes Asd and DapA, and DapA and DapB in the L-lysine fermentation pathway through ligand peptides, respectively, and linked to plasmid expression vectors. The recombinant plasmid was transferred to L-lysine-producing bacterium E. coli QDE, and two strains of intracellular pair-assembled engineered bacteria, QDE::pET-28a(+)-asd-Coh/DocA-S3-dapA and QDE::pET-28a(+)-dapA-Coh/DocA-S3-dapB, were obtained.
The cumulative L-lysine concentration of the assembled engineered strain QDE::pET-28a(+)-
asd-Coh /DocA-S3-
dapA reached 58.0 g/L, 14.8% higher than that of the control strain QDE::pET-28a(+)-
asd/dapA, as shown in
Figure 10. The cumulative L-lysine concentration of the engineered QDE::pET-28a(+) -
dapA-Coh /DocA-S3-
dapB reached 55.3 g/L, 29.8% higher than that of the control strain QDE::pET-28a(+)-
dapA/dapB, as shown in
Figure 11. This strategy can exert the “proximity effect” among multi-enzyme complexes, improve the transfer efficiency of intermediate metabolites between different catalytic active centers, and indirectly improve the catalytic rate of each key enzyme.
Based on the above verification, the use of DocA-S3 components improved the L-lysine fermentation efficiency of the production strains significantly. Therefore, DocA-S3 could be used to assemble intracellular complexes and improve their synthetic efficiency.
4. Conclusions
To address the strong extracellular Ca2+ dependence problem in the assembly of DocA and Coh proteins, this study conducted site-specific studies on the key sites of Ca2+ binding in DocA, and finally obtained three Ca2+ independent DocA mutants.
After constructing the corresponding engineering strains, the extracellular expression of purified protein was detected in a low Ca2+ environment, and the mutant DocA-S3 had the strongest binding ability with Coh, which was 3.8–4.6 times that of DocA. The fluorescence signal intensity of the fusion protein assembled with DocA-S3 was as high as 1.50 × 107 in E. coli cells, which was significantly improved compared with that of the initial DocA, indicating that the mutant could interact with Coh in vivo. Molecular dynamics simulations and conformational analysis showed that in the absence of Ca2+ addition, the D5R mutation in DocA-S3 could form a new hydrogen bond network with Gly and Val, ensuring that the double α-helix maintained the stability of its interaction with Coh at a certain angle and formed an intracellular self-assembly complex independent of Ca2+.
The precise regulatory mechanism of the application of DocA-S3 to the assembly of key enzymes for L-lysine biosynthesis still needs to be studied further. In the present study, the intracellular assembly of Asd and DapA, DapA and DapB was realized, laying a foundation for the design of key enzymes for L-lysine sequential reaction in intracellular assembly. It provides a new strategy for improving intracellular cellulosome self-assembly and amino acid fermentation efficiency.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}