
Unraveling the Complexity of Vaso-Occlusive Crises in Sickle Cell Disease: Insights from a Resource-Limited Setting
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Environment
2.2. Population
2.3. Data Collection
2.4. Statistical Analysis
3. Results
3.1. Patients and VOCs
3.2. Complications
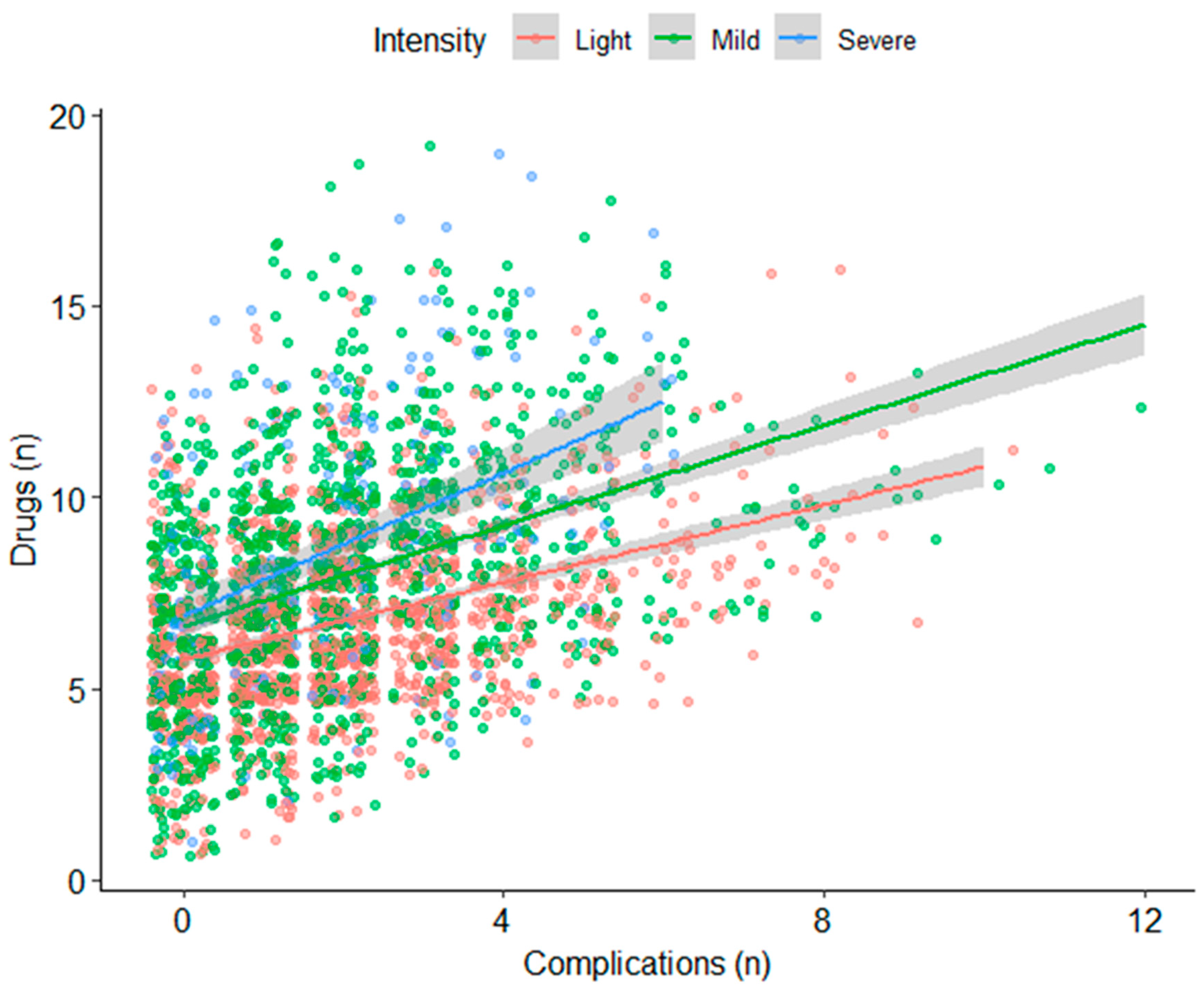
3.3. Medications
3.4. Biology
3.5. Survival Analysis
4. Discussion
4.1. Demographic Data
4.2. VOC’s Type
4.3. Infections
4.4. Medications
4.5. Biology
4.6. Predictions of VOCs
4.7. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- John, N. A Review of Clinical Profile in Sickle Cell Traits. Oman Med. J. 2010, 25, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global Burden of Sickle Cell Anaemia in Children under Five, 2010–2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle Cell Disease. Nat. Rev. Dis. Primer 2018, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Vinjamur, D.S.; Bauer, D.E.; Orkin, S.H. Recent Progress in Understanding and Manipulating Haemoglobin Switching for the Haemoglobinopathies. Br. J. Haematol. 2018, 180, 630–643. [Google Scholar] [CrossRef]
- Ballas, S.K.; Darbari, D.S. Review/Overview of Pain in Sickle Cell Disease. Complement. Ther. Med. 2020, 49, 102327. [Google Scholar] [CrossRef]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-Cell Disease. Lancet Lond. Engl. 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Darbari, D.S.; Sheehan, V.A.; Ballas, S.K. The Vaso-Occlusive Pain Crisis in Sickle Cell Disease: Definition, Pathophysiology, and Management. Eur. J. Haematol. 2020, 105, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Solovey, A.; Kollander, R.; Milbauer, L.C.; Abdulla, F.; Chen, Y.; Kelm, R.J.; Hebbel, R.P. Endothelial Nitric Oxide Synthase and Nitric Oxide Regulate Endothelial Tissue Factor Expression in Vivo in the Sickle Transgenic Mouse. Am. J. Hematol. 2010, 85, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Manwani, D.; Frenette, P.S. Vaso-Occlusion in Sickle Cell Disease: Pathophysiology and Novel Targeted Therapies. Blood 2013, 122, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K. Sickle Cell Disease: Classification of Clinical Complications and Approaches to Preventive and Therapeutic Management. Clin. Hemorheol. Microcirc. 2018, 68, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Shanley, L.A.; Ebeling, M.; Titus, M.O. Changes in Platelet Count as a Predictive Tool in Sickle Cell Acute Vaso-Occlusive Crises: A Pediatric Study. Clin. Pediatr. 2011, 50, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Darbari, D.S.; Onyekwere, O.; Nouraie, M.; Minniti, C.P.; Luchtman-Jones, L.; Rana, S.; Sable, C.; Ensing, G.; Dham, N.; Campbell, A.; et al. Markers of Severe Vaso-Occlusive Painful Episode Frequency in Children and Adolescents with Sickle Cell Anemia. J. Pediatr. 2012, 160, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Stankovic Stojanovic, K.; Steichen, O.; Lefevre, G.; Bachmeyer, C.; Avellino, V.; Grateau, G.; Girot, R.; Lionnet, F. High Lactate Dehydrogenase Levels at Admission for Painful Vaso-Occlusive Crisis Is Associated with Severe Outcome in Adult SCD Patients. Clin. Biochem. 2012, 45, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Ugwu, A.O.; Ibegbulam, O.G.; Nwagha, T.U.; Madu, A.J.; Ocheni, S.; Okpala, I. Clinical and Laboratory Predictors of Frequency of Painful Crises among Sickle Cell Anaemia Patients in Nigeria. J. Clin. Diagn. Res. JCDR 2017, 11, EC22–EC25. [Google Scholar] [CrossRef] [PubMed]
- Egesa, W.I.; Nakalema, G.; Waibi, W.M.; Turyasiima, M.; Amuje, E.; Kiconco, G.; Odoch, S.; Kumbakulu, P.K.; Abdirashid, S.; Asiimwe, D. Sickle Cell Disease in Children and Adolescents: A Review of the Historical, Clinical, and Public Health Perspective of Sub-Saharan Africa and Beyond. Int. J. Pediatr. 2022, 2022, 3885979. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, A.A.F.; Ribeiro, S.B.F.; Moraes-Souza, H.; de Oliveira, L.F.; Ribeiro, J.B.; da Silva, S.H.; de Oliveira, D.F.F.; Ribeiro, M.F.F. Pain Measurement as Part of Primary Healthcare of Adult Patients with Sickle Cell Disease. Rev. Bras. Hematol. Hemoter. 2013, 35, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Zempsky, W.T.; Campbell-Yeo, M.; Chambers, C.T.; Cohen, L.L.; Gagliese, L.; Kwok, C.H.T.; Trang, T.; Stevens, B.; Taddio, A.; Voepel-Lewis, T.; et al. Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks-American Pain Society-American Academy of Pain Medicine Pain Taxonomy Diagnostic Criteria for Acute Needle Pain. J. Pain 2023, 24, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Serjeant, G.R. The Natural History of Sickle Cell Disease. Cold Spring Harb. Perspect. Med. 2013, 3, a011783. [Google Scholar] [CrossRef]
- McClish, D.K.; Smith, W.R.; Dahman, B.A.; Levenson, J.L.; Roberts, J.D.; Penberthy, L.T.; Aisiku, I.P.; Roseff, S.D.; Bovbjerg, V.E. Pain Site Frequency and Location in Sickle Cell Disease: The PiSCES Project. Pain 2009, 145, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Dembélé, A.K.; Toure, B.A.; Sarro, Y.S.; Guindo, A.; Fané, B.; Offredo, L.; Kené, S.; Conaré, I.; Tessougué, O.; Traoré, Y.; et al. Prevalence and risk factors for sickle retinopathy in a sub-Saharan comprehensive Sickle Cell Center. Rev. Med. Interne 2017, 38, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J.; Malik, P.; Vercellotti, G.M. Therapeutic Strategies for Sickle Cell Disease: Towards a Multi-Agent Approach. Nat. Rev. Drug Discov. 2019, 18, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Spira, J.A.O.; Borges, E.L.; Guedes, A.C.M.; Andrade, P.G.R.; de Araujo Nogueira Lima, V.L. Prevalence of People with Sickle Cell Disease and Leg Ulcers in Brazil: Socioeconomic and Clinical Overview. PLoS ONE 2022, 17, e0274254. [Google Scholar] [CrossRef] [PubMed]
- Estcourt, L.J.; Fortin, P.M.; Hopewell, S.; Trivella, M.; Wang, W.C. Blood Transfusion for Preventing Primary and Secondary Stroke in People with Sickle Cell Disease. Cochrane Database Syst. Rev. 2017, 1, CD003146. [Google Scholar] [CrossRef] [PubMed]
- Frediani, J.K.; Pak-Harvey, E.; Parsons, R.; Westbrook, A.L.; O’Sick, W.; Martin, G.S.; Lam, W.A.; Levy, J.M. Prevalence of SARS-CoV-2 in Hemoglobinopathies Is Modified by Age and Race. Blood Cells. Mol. Dis. 2023, 102, 102756. [Google Scholar] [CrossRef] [PubMed]
- Udeze, C.; Evans, K.A.; Yang, Y.; Lillehaugen, T.; Manjelievskaia, J.; Mujumdar, U.; Li, N.; Andemariam, B. Economic and Clinical Burden of Managing Sickle Cell Disease with Recurrent Vaso-Occlusive Crises in the United States. Adv. Ther. 2023, 40, 3543–3558. [Google Scholar] [CrossRef] [PubMed]
- Platt, O.S.; Thorington, B.D.; Brambilla, D.J.; Milner, P.F.; Rosse, W.F.; Vichinsky, E.; Kinney, T.R. Pain in Sickle Cell Disease. Rates and Risk Factors. N. Engl. J. Med. 1991, 325, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; Lobo, C.; Queiroz, A.M.M.; do Nascimento, E.M.; Lima, C.B.; Cardoso, G.; Ballas, S.K. Treatment of the Acute Sickle Cell Vaso-Occlusive Crisis in the Emergency Department: A Brazilian Method of Switching from Intravenous to Oral Morphine. Eur. J. Haematol. 2014, 93, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.R.; Scherer, M. Sickle-Cell Pain: Advances in Epidemiology and Etiology. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Many, M.; Mashako, Y.; Bitwe, M.; Nsibu, C. Profil-Épidémiologique-et-Clinique-de-La-Drépanocytose-à-l’hôpital-Provincial-Du-Nord-Kivu.-OK. Rev. Malg. Ped. 2020, 2, 62–69. [Google Scholar]
- Arlet, J.-B.; Cheminet, G.; Allali, S. Les Crises Vaso-Occlusives de La Drépanocytose. Presse Méd. Form. 2021, 2, 373–379. [Google Scholar] [CrossRef]
- Claeys, A.; Van Steijn, S.; Van Kesteren, L.; Damen, E.; Van Den Akker, M. Varied Age of First Presentation of Sickle Cell Disease: Case Presentations and Review. Case Rep. Med. 2021, 2021, 8895020. [Google Scholar] [CrossRef] [PubMed]
- John, C.C.; Opoka, R.O.; Latham, T.S.; Hume, H.A.; Nabaggala, C.; Kasirye, P.; Ndugwa, C.M.; Lane, A.; Ware, R.E. Hydroxyurea Dose Escalation for Sickle Cell Anemia in Sub-Saharan Africa. N. Engl. J. Med. 2020, 382, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Cehemegni, B.C. Complications aigües de la drépanocytose aiguës. J. Maroc. Sci. Méd. 2017, 21, 18–23. [Google Scholar] [CrossRef]
- Landouré, G.; Cissé, L.; Touré, B.A.; Yalcouyé, A.; Coulibaly, T.; Karambé, M.; Sissoko, A.S.; Coulibaly, T.; Wonkam, A.; Guinto, C.O.; et al. Neurological Complications in Subjects With Sickle Cell Disease or Trait: Genetic Results from Mali. Glob. Heart 2017, 12, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Diop, M.M.; Camara, E.; Barry, I.K.; Barry, A.; Doukoure, M.A.; Barry, M.C.; Diallo, M.L.; Kouyate, M.; Diallo, S.B.; Camara, D.D.; et al. Profil Clinique et Biologique des Drépanocytaires sous Hydroxyurée au Service de Pédiatrie de l’Hôpital National Donka (Conakry). Health Sci. Dis. 2020, 21, 75–77. [Google Scholar]
- Forni, G.L.; Finco, G.; Graziadei, G.; Balocco, M.; Rigano, P.; Perrotta, S.; Olivieri, O.; Cappellini, M.D.; De Franceschi, L. Development of Interactive Algorithm for Clinical Management of Acute Events Related to Sickle Cell Disease in Emergency Department. Orphanet J. Rare Dis. 2014, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.J.; Kopecky, E.A.; Joshi, P.; Babul, N. Randomised Trial of Oral Morphine for Painful Episodes of Sickle-Cell Disease in Children. Lancet Lond. Engl. 1997, 350, 1358–1361. [Google Scholar] [CrossRef] [PubMed]
- Alsalman, M.; Alhabrati, A.; Alkuwaiti, A.; Alramadhan, N.; AlMurayhil, N.; Althafar, A.; Alsaad, A. Depression among Patient with Sickle Cell Disease: Prevalence and Prediction. Niger. J. Clin. Pract. 2022, 25, 1274–1278. [Google Scholar] [CrossRef]
- Mishkin, A.D.; Prince, E.J.; Leimbach, E.J.; Mapara, M.Y.; Carroll, C.P. Psychiatric Comorbidities in Adults with Sickle Cell Disease: A Narrative Review. Br. J. Haematol. 2023, 203, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Dae, A.; Km, G.; Yd, A.; Ke, D.; Tsolenyanu, E.; Ad, G.; Yd, A. Prise En Charge de La Douleur Lors Des Crises Vaso-Occlusives Sévères de l’Enfant Au CHU Sylvanus Olympio de Lomé (Togo). Health Sci. Dis. 2023, 2, 10–14. [Google Scholar]
- Cintron-Garcia, J.; Ajebo, G.; Kota, V.; Guddati, A.K. Mortality Trends in Sickle Cell Patients. Am. J. Blood Res. 2020, 10, 190–197. [Google Scholar] [PubMed]
- Ramsey, S.D.; Bender, M.A.; Li, L.; Johnson, K.M.; Jiao, B.; Devine, B.; Basu, A. Prevalence of Comorbidities Associated with Sickle Cell Disease among Non-Elderly Individuals with Commercial Insurance-A Retrospective Cohort Study. PLoS ONE 2022, 17, e0278137. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Bhor, M.; Xie, L.; Paulose, J.; Yuce, H. Sickle Cell Disease Complications: Prevalence and Resource Utilization. PLoS ONE 2019, 14, e0214355. [Google Scholar] [CrossRef] [PubMed]
- Falletta, J.M.; Woods, G.M.; Verter, J.I.; Buchanan, G.R.; Pegelow, C.H.; Iyer, R.V.; Miller, S.T.; Holbrook, C.T.; Kinney, T.R.; Vichinsky, E. Discontinuing Penicillin Prophylaxis in Children with Sickle Cell Anemia. Prophylactic Penicillin Study II. J. Pediatr. 1995, 127, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Gaston, M.H.; Verter, J.I.; Woods, G.; Pegelow, C.; Kelleher, J.; Presbury, G.; Zarkowsky, H.; Vichinsky, E.; Iyer, R.; Lobel, J.S. Prophylaxis with Oral Penicillin in Children with Sickle Cell Anemia. A Randomized Trial. N. Engl. J. Med. 1986, 314, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.T.; Tshilolo, L.; Santos, B.; Tomlinson, G.A.; Stuber, S.; Latham, T.; Aygun, B.; Obaro, S.K.; Olupot-Olupot, P.; Williams, T.N.; et al. Hydroxyurea Therapy for Children with Sickle Cell Anemia in Sub-Saharan Africa: Rationale and Design of the REACH Trial. Pediatr. Blood Cancer 2016, 63, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; Barton, F.B.; Eckert, S.V.; McMahon, R.P.; Bonds, D.R. Effect of Hydroxyurea on the Frequency of Painful Crises in Sickle Cell Anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Yawn, B.P.; John-Sowah, J. Management of Sickle Cell Disease: Recommendations from the 2014 Expert Panel Report. Am. Fam. Physician 2015, 92, 1069–1076. [Google Scholar] [PubMed]
- Ware, R.E.; De Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle Cell Disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Olupot-Olupot, P.; Connon, R.; Kiguli, S.; Opoka, R.O.; Alaroker, F.; Uyoga, S.; Nakuya, M.; Okiror, W.; Nteziyaremye, J.; Ssenyondo, T.; et al. A Predictive Algorithm for Identifying Children with Sickle Cell Anemia among Children Admitted to Hospital with Severe Anemia in Africa. Am. J. Hematol. 2022, 97, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Mukinayi, B.M.; Cibeyibeyi, G.K.; Tumba, G.D.; Gulbis, B. Drépanocytose En République Démocratique Du Congo: Quels Sont Les Obstacles à Un Traitement Par Hydroxyurée? Pan Afr. Med. J. 2021, 38, 41. [Google Scholar] [CrossRef] [PubMed]
- Mathilde, B.; Alex, B.I.; Jolie, B.M.; Masanka, K.; Philomène; Alain, A.; Simon, K.; Michel, K.; Panda; Jules, M.; et al. Availability and Cost of Hydroxyuera Used in the Management of Sickle Cell Disease in Lubumbashi, Democratic Republic of Congo. Int. J. Curr. Res. Life Sci. 2018, 7, 2336–2340. [Google Scholar]
- Kamble, M.; Chatruvedi, P. Epidemiology of Sickle Cell Disease in a Rural Hospital of Central India. Indian Pediatr. 2000, 37, 391–396. [Google Scholar] [PubMed]
- Tluway, F.; Makani, J. Sickle Cell Disease in Africa: An Overview of the Integrated Approach to Health, Research, Education and Advocacy in Tanzania, 2004-2016. Br. J. Haematol. 2017, 177, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in Sickle Cell Disease. Life Expectancy and Risk Factors for Early Death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Hsu, H.; Nagel, R.L.; Milner, P.F.; Adams, J.G.; Benjamin, L.; Fryd, S.; Gillette, P.; Gilman, J.; Josifovska, O. Gender and Haplotype Effects upon Hematological Manifestations of Adult Sickle Cell Anemia. Am. J. Hematol. 1995, 48, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Diallo, D.A. Sickle cell disease in Africa: Current situation and strategies for improving the quality and duration of survival. Bull. Acad. Natl. Med. 2008, 192, 1361–1372; discussion 1372–1373. [Google Scholar] [PubMed]
- Diallo, D.A.; Baby, M.; Boiré, A.; Diallo, Y.L. Management of pain of acute sickle cell pain crises by health care providers in Mali. Med. Trop. Rev. Corps Sante Colon. 2008, 68, 502–506. [Google Scholar]
- Baye, D.; Belemou, B.; Dapa, D.; Dembele Keita, H.; Marouf, M.; Ouane, O.D.; Sangho, H.; Sidibe Keita, A.; Sidibe, T. Enquete CAP des agents de sante sur la prise en charge de l’enfant drepanocytaire a Bamako. Mali Méd. En Ligne 2008, 23, 1–4. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Severity | Localization | ||
|---|---|---|---|
| Osteoarticular | Mixed | Abdominal | |
| Light | n = 694 Age = 9.0 [11] Sex ratio = 51.1% | n = 337 Age = 9.8 [10] Sex ratio = 52.6% | n = 285 Age = 8.7 [8] Sex ratio = 57.6% |
| Mild | n = 872 Age = 11.3 [13] Sex ratio = 55.3% | n = 398 Age = 9.6 [13] Sex ratio = 62.9% | n = 90 Age = 17.6 [12] Sex ratio = 58.3% |
| Severe | n = 164 Age = 13.7 [8] Sex ratio = 61.2% | n = 58 Age = 9.2 [9] Sex ratio = 52.9% | n = 12 Age = 6.2 [2] Sex ratio = 33.3% |
| Complications | Overall (n = 2910) | Severity | ||
|---|---|---|---|---|
| Light (n = 1316) | Mild (n = 1360) | Severe (n = 234) | ||
| Articular | 2.7% | 2.1% | 3.3% | 2.1% |
| Digestif | 43.7% | 47.3% | 40.2% | 37.3% |
| Ear, Nose, and Throat | 17.5% | 23.0% | 14.3% | 4.5% |
| Para-infectious | 86.2% | 86.3% | 86.6% | 74.9% |
| Musculo-cutaneous | 2.5% | 3.3% | 2.0% | 1.2% |
| Blood | 18.2% | 10.2% | 22.1% | 37.5% |
| Cardiovascular | 2.7% | 2.1% | 3.3% | 2.1% |
| Respiratory | 25.7% | 28.0% | 24.3% | 19.3% |
| Gynecologic | 0.6% | 0.5% | 0.6% | 0.4% |
| Medications (Family) | Overall (n = 2910) | Severity | ||
|---|---|---|---|---|
| Light (n = 1316) | Mild (n = 1360) | Severe (n = 234) | ||
| Amino acids (AAs) | 9.1% | 3.8% | 11.5% | 24.8% |
| Analgesic-anti-inflammatory agents (AAIs) | 1.4% | 0.8% | 2.0% | 1.7% |
| Antacids and proton pump inhibitors (AAINPs) | 14.4% | 9.5% | 17.4% | 24.8% |
| Anticoagulants (ACOs) | 1.1% | 1.1% | 1.1% | 0.9% |
| Antiemetic (AEM) | 7.4% | 3.2% | 10.0% | 15.4% |
| Anti-inflammatory (AI) | 152.3% | 105.6% | 188.9% | 202.6% |
| Antifungals (ANFs) | 2.1% | 2.3% | 2.0% | 1.3% |
| Antioxidants (ANOs) | 23.5% | 29.8% | 19.1% | 13.7% |
| Antiparasitics (ANPs) | 80.6% | 95.6% | 70.1% | 56.8% |
| Analgesics (ANTs) | 69.7% | 26.4% | 101.1% | 130.3% |
| Analgesic-antipyretic (ANTP) | 78.1% | 62.9% | 89.3% | 98.3% |
| Antispasmodics (ASMs) | 27.4% | 26.9% | 28.2% | 25.2% |
| Antibiotics (ATBs) | 81.1% | 87.2% | 76.8% | 72.6% |
| Antibiotics and antiparasitics (ATB-ATPs) | 13.4% | 15.3% | 12.1% | 9.8% |
| Antihistamines (ATHs) | 4.4% | 5.2% | 4.0% | 1.3% |
| Antiseptics (ATSs) | 1.1% | 1.4% | 0.9% | 0.4% |
| Antitussives (ATTs) | 17.8% | 22.9% | 14.3% | 9.8% |
| Nasal decongestants (Cos) | 3.3% | 4.7% | 2.4% | 0.9% |
| Diuretics (DIURs) | 0.8% | 0.3% | 1.2% | 1.3% |
| Hydrating agents (HYDs) | 19.8% | 13.7% | 20.4% | 51.3% |
| Hemoglobin inducers and antioxidants (IhbAOs) | 21.6% | 20.5% | 21.8% | 26.5% |
| Fetal hemoglobin inducers and antioxidants (IHBFAOs) | 7.1% | 8.7% | 5.8% | 6.0% |
| Minerals (MINs) | 6.1% | 7.1% | 5.6% | 3.8% |
| Gastric dressing (PG) | 23.0% | 25.2% | 21.7% | 17.9% |
| Vasodilators (VDCs) | 4.3% | 2.4% | 3.9% | 17.1% |
| Vitamins (VITs) | 70.3% | 83.1% | 59.2% | 62.8% |
| Vitamins-minerals (VIT-MINs) | 5.7% | 5.2% | 6.0% | 6.0% |
| Parameters | n | Beta (Complications) | p-Value |
|---|---|---|---|
| Hemoglobin | 1454 | 0.18 (0.04) | 9.14 × 106 |
| Sedimentation rate | 896 | 0.78 (0.15) | 4.55 × 107 |
| Leucocytes | 978 | 517 (88) | 5.84 × 109 |
| Lymphocytes | 889 | 0.74 (0.22) | 7.24 × 104 |
| Monocytes | 442 | 0.03 (0.02) | 0.30 |
| Neutrophils | 888 | 1.13 (0.26) | 2.17 × 105 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaponda, A.; Muya, K.; Panda, J.; Koto, K.K.; Bonnechère, B. Unraveling the Complexity of Vaso-Occlusive Crises in Sickle Cell Disease: Insights from a Resource-Limited Setting. J. Clin. Med. 2024, 13, 2528. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm13092528
Kaponda A, Muya K, Panda J, Koto KK, Bonnechère B. Unraveling the Complexity of Vaso-Occlusive Crises in Sickle Cell Disease: Insights from a Resource-Limited Setting. Journal of Clinical Medicine. 2024; 13(9):2528. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm13092528
Chicago/Turabian StyleKaponda, Ali, Kalunga Muya, Jules Panda, Kodondi Kule Koto, and Bruno Bonnechère. 2024. "Unraveling the Complexity of Vaso-Occlusive Crises in Sickle Cell Disease: Insights from a Resource-Limited Setting" Journal of Clinical Medicine 13, no. 9: 2528. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm13092528