
A Novel System for the Detection of Spontaneous Abortion-Causing Aneuploidy and Its Erroneous Chromosome Origins through the Combination of Low-Pass Copy Number Variation Sequencing and NGS-Based STR Tests
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. DNA Extraction and CNV-Seq
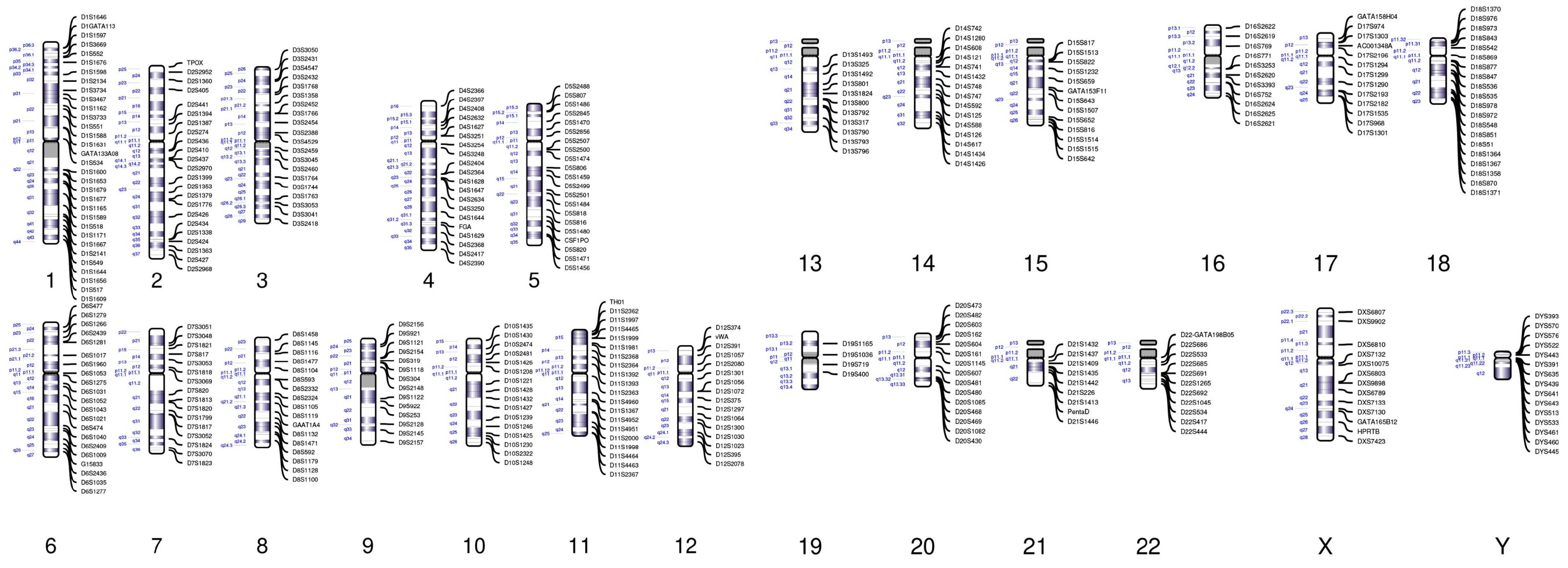
2.3. NGS-Based STR Test
2.4. Chromosome G-Banding Analysis
3. Results
3.1. Construction of an NGS-Based STR Panel
3.2. CNV-Seq Combined with the STR Panel Analysis of the Results of Double Trisomy
3.3. CNV-Seq Combined with the STR Panel Analysis of the Results of UDP
3.4. CNV-Seq Combined with the STR Panel Analysis of the Results of Triploidy
3.5. Results of CNV-Seq Combined with the STR Panel of 500 Unexplained RSA Miscarriage Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassold, T.; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet. 2001, 2, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Jurkovic, D.; Overton, C.; Bender-Atik, R. Diagnosis and management of first trimester miscarriage. BMJ 2013, 346, f3676. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.; Dise, C.A.; Benito, C.W.; Ziadie, M.S.; Hovanes, K. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: Outcomes, benefits, and challenges. Genet. Med. 2017, 19, 83–89. [Google Scholar] [CrossRef]
- Al-Memar, M.; Bobdiwala, S.; Fourie, H.; Mannino, R.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Timmerman, D.; Bourne, T.; Bennett, P.R.; et al. The association between vaginal bacterial composition and miscarriage: A nested case-control study. BJOG 2022, 127, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Amrane, S.; McConnell, R. Endocrine causes of recurrent pregnancy loss. Semin. Perinatol. 2019, 43, 80–83. [Google Scholar] [CrossRef]
- Begtrup, L.M.; Specht, I.O.; Hammer, P.E.C.; Flachs, E.M.; Garde, A.H.; Hansen, J.; Hansen, Å.M.; Kolstad, H.A.; Larsen, A.D.; Bonde, J.P. Night work and miscarriage: A Danish nationwide register-based cohort study. Occup. Environ. Med. 2019, 76, 302–308. [Google Scholar] [CrossRef]
- Musizzano, Y.; Fulcheri, E. Decidual vascular patterns in first-trimester abortions. Virchows Arch. 2010, 456, 543–560. [Google Scholar] [CrossRef]
- Whittaker, P.G.; Schreiber, C.A.; Sammel, M.D. Gestational hormone trajectories and early pregnancy failure: A reassessment. Reprod. Biol. Endocrinol. 2018, 16, 95. [Google Scholar] [CrossRef]
- Colley, E.; Hamilton, S.; Smith, P.; Morgan, N.V.; Coomarasamy, A.; Allen, S. Potential genetic causes of miscarriage in euploid pregnancies: A systematic review. Hum. Reprod. Update 2019, 25, 452–472. [Google Scholar] [CrossRef]
- Gug, C.; Rațiu, A.; Navolan, D.; Drăgan, I.; Groza, I.M.; Păpurică, M.; Vaida, M.A.; Mozoș, I.; Jurcă, M.C. Incidence and Spectrum of Chromosome Abnormalities in Miscarriage Samples: A Retrospective Study of 330 Cases. Cytogenet. Genome Res. 2019, 158, 171–183. [Google Scholar] [CrossRef]
- Yakut, S.; Toru, H.S.; Çetin, Z.; Özel, D.; Şimşek, M.; Mendilcioğlu, İ.; Lüleci, G. Chromosome abnormalities identified in 457 spontaneous abortions and their histopathological findings. Turk. Patoloji. Derg. 2015, 31, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, M.; Kutteh, W. Evaluation and management of recurrent early pregnancy loss. Clin. Obstet. Gynecol. 2007, 50, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Suzumori, N.; Sugiura-Ogasawara, M. Genetic factors as a cause of miscarriage. Curr. Med. Chem. 2010, 17, 3431–3437. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Chen, L.; Lin, J.; Zhu, J.; Zhang, N.; Qiu, X.; Li, D.; Wang, L. Chromosomal karyotype in chorionic villi of recurrent spontaneous abortion patients. Biosci. Trends 2018, 12, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Portilla, R.J.; Pauta, M.; Hawkins-Villarreal, A.; Rial-Crestelo, M.; Paz, Y.M.F.; Madrigal, I.; Figueras, F.; Borrell, A. Added value of chromosomal microarray analysis over conventional karyotyping in stillbirth work-up: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2019, 53, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.C.; Chow, J.F.; Lau, E.Y.; Yeung, W.S.; Ho, P.C.; Ng, E.H. Comparison between fluorescent in-situ hybridisation and array comparative genomic hybridisation in preimplantation genetic diagnosis in translocation carriers. Hong Kong Med. J. 2015, 21, 16–22. [Google Scholar] [CrossRef]
- Lescoat, D.; Jouan, H.; Loeuillet-Olivo, L.; Le Calvé, M. Fluorescent in situ hybridization (FISH) on paraffin-embedded placental tissues as an adjunct for understanding the etiology of early spontaneous abortion. Prenat. Diagn. 2005, 25, 314–317. [Google Scholar] [CrossRef]
- Qu, S.; Wang, L.; Cai, A.; Bai, N.; Liu, N.; Kong, X. Exploring the cause of early miscarriage with SNP-array analysis and karyotyping. J. Matern. Fetal Neonatal Med. 2019, 32, 1–10. [Google Scholar] [CrossRef]
- Ramasamy, R.; Scovell, J.M.; Kovac, J.R.; Cook, P.J.; Lamb, D.J.; Lipshultz, L.I. Fluorescence in situ hybridization detects increased sperm aneuploidy in men with recurrent pregnancy loss. Fertil. Steril. 2015, 103, 906–909.e1. [Google Scholar] [CrossRef] [Green Version]
- Erol, E.; Jackson, C.B.; Steinman, M.; Meares, K.; Donahoe, J.; Kelly, N.; Locke, S.; Smith, J.L.; Carter, C.N. A diagnostic evaluation of real-time PCR, fluorescent antibody and microscopic agglutination tests in cases of equine leptospiral abortion. Equine Vet. J. 2015, 47, 171–174. [Google Scholar] [CrossRef]
- Kim, J.W.; Lyu, S.W.; Sung, S.R.; Park, J.E.; Cha, D.H.; Yoon, T.K.; Ko, J.J.; Shim, S.H. Molecular analysis of miscarriage products using multiplex ligation-dependent probe amplification (MLPA): Alternative to conventional karyotype analysis. Arch. Gynecol. Obstet. 2015, 291, 347–354. [Google Scholar] [CrossRef]
- Zimowski, J.G.; Massalska, D.; Pawelec, M.; Bijok, J.; Michałowska, A.; Roszkowski, T. First-trimester spontaneous pregnancy loss—Molecular analysis using multiplex ligation-dependent probe amplification. Clin. Genet. 2016, 89, 620–624. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Chen, X.; Wang, D.; Chen, X.; Wang, C.; Zhang, Y.; Xu, M.; Yu, J. Prevalence of chromosomal abnormalities identified by copy number variation sequencing in high-risk pregnancies, spontaneous abortions, and suspected genetic disorders. J. Int. Med. Res. 2019, 47, 1169–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Song, L.; Cram, D.S.; Xiong, L.; Wang, K.; Wu, R.; Liu, J.; Deng, K.; Jia, B.; Zhong, M.; et al. Traditional karyotyping vs. copy number variation sequencing for detection of chromosomal abnormalities associated with spontaneous miscarriage. Ultrasound Obstet. Gynecol. 2005, 46, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cram, D.S.; Shen, J.; Wang, X.; Zhang, J.; Song, Z.; Xu, G.; Li, N.; Fan, J.; Wang, S.; et al. Validation of copy number variation sequencing for detecting chromosome imbalances in human preimplantation embryos. Biol. Reprod. 2014, 91, 37. [Google Scholar] [CrossRef] [PubMed]
- Amunugama, R.; Fishel, R. Homologous recombination in eukaryotes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 155–206. [Google Scholar] [CrossRef]
- Crickard, J.B.; Greene, E.C. The biochemistry of early meiotic recombination intermediates. Cell Cycle 2018, 17, 2520–2530. [Google Scholar] [CrossRef] [Green Version]
- Ottolini, C.S.; Newnham, L.; Capalbo, A.; Natesan, S.A.; Joshi, H.A.; Cimadomo, D.; Housworth, E.; Herbert, A.D.; Rienzi, L.; Ubaldi, F.M.; et al. Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat. Genet. 2015, 47, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Pratto, F.; Brick, K.; Khil, P.; Smagulova, F.; Petukhova, G.V.; Camerini-Otero, R.D. Recombination initiation maps of individual human genomes. Science 2014, 346, 1256442. [Google Scholar] [CrossRef] [Green Version]
- de la Seña, C.A.; Fechheimer, N.S.; Nestor, K.E. Evidence for genetic etiology of heteroploidy in embryos of the Japanese quail (Coturnix coturnix japonica). Cytogenet. Cell Genet. 1992, 60, 140–145. [Google Scholar] [CrossRef]
- Jacobs, P.A.; Angell, R.R.; Buchanan, I.M.; Hassold, T.J.; Matsuyama, A.M.; Manuel, B. The origin of human triploids. Ann. Hum. Genet. 1978, 42, 49–57. [Google Scholar] [CrossRef]
- Robinson, W.P.; McFadden, D.E.; Stephenson, M.D. The origin of abnormalities in recurrent aneuploidy/polyploidy. Am. J. Hum. Genet. 2001, 69, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Bug, S.; Solfrank, B.; Schmitz, F.; Pricelius, J.; Stecher, M.; Craig, A.; Botcherby, M.; Nevinny-Stickel-Hinzpeter, C. Diagnostic utility of novel combined arrays for genome-wide simultaneous detection of aneuploidy and uniparental isodisomy in losses of pregnancy. Mol. Cytogenet. 2014, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Sebire, N.J.; May, P.C.; Kaur, B.; Seckl, M.J.; Fisher, R.A. Abnormal villous morphology mimicking a hydatidiform mole associated with paternal trisomy of chromosomes 3, 7, 8 and unipaternal disomy of chromosome 11. Diagn. Pathol. 2016, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Ercan, F.; Taşdemir, P.; Tazegül Pekin, A.; Sayal, B.; Görkemli, H.; Acar, A. Prenatal diagnosis of double trisomy 48, XXX, +18; case report. J. Obstet. Gynaecol. 2018, 38, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Wu, X.H.; Zou, C.C. Double trisomy 48, XXX, +18 with multiple dysmorphic features. World J. Pediatr. 2015, 11, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.S. Double trisomy in spontaneous abortions. Hum. Genet. 1997, 101, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.I.; Dyer, L.; Stanek, J. Placental Histomorphology in a Case of Double Trisomy 48, XXX, +18. Case Rep. Pathol. 2018, 2018, 2839765. [Google Scholar] [CrossRef] [Green Version]
- Vergara-Mendez, L.D.; Talero-Gutiérrez, C.; Velez-Van-Meerbeke, A. Double trisomy (XXX+21 karyotype) in a six-year-old girl with down phenotype. J. Genet. 2018, 97, 337–340. [Google Scholar] [CrossRef]
- Watabe, T.; Koga, H. Survival in double aneuploidy involving trisomy 18 and sex chromosome trisomy: A case report of a 27-month-old child and a review of the literature. Congenit. Anom. 2019, 59, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hassed, S.; Mulvihill, J.J.; Nair, A.K.; Hopcus, D.J. Double trisomy. Am. J. Med. Genet. A 2004, 124A, 96–98. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Count | Ratio in Total Samples | Ratio in Abnormal Samples | |
|---|---|---|---|
| Trisomy | 169 | 33.4% | 59.9% |
| Double trisomy | 6 | 1.2% | 2.1% |
| Triploid | 40 | 8.0% | 14.2% |
| Monosomy | 32 | 6.4% | 11.3% |
| UPD | 16 | 3.2% | 5.7% |
| Chromosome segmental microduplication or microdeletion | 18 | 3.6% | 6.4% |
| Monosomy + trisomy | 1 | 0.2% | 0.4% |
| Maternal contamination | 2 | 0.4% | / |
| Normal | 216 | 43.2% | / |
| Total | 500 | ||
| Abnormal total | 282 | 56.4% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, C.; Liao, K.; Zhao, Y.; Long, Z.; Zhu, S.; Wu, J.; Xiao, M.; Zhou, J.; Zhang, S.; Li, L.; et al. A Novel System for the Detection of Spontaneous Abortion-Causing Aneuploidy and Its Erroneous Chromosome Origins through the Combination of Low-Pass Copy Number Variation Sequencing and NGS-Based STR Tests. J. Clin. Med. 2023, 12, 1809. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12051809
Lei C, Liao K, Zhao Y, Long Z, Zhu S, Wu J, Xiao M, Zhou J, Zhang S, Li L, et al. A Novel System for the Detection of Spontaneous Abortion-Causing Aneuploidy and Its Erroneous Chromosome Origins through the Combination of Low-Pass Copy Number Variation Sequencing and NGS-Based STR Tests. Journal of Clinical Medicine. 2023; 12(5):1809. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12051809
Chicago/Turabian StyleLei, Caixia, Kai Liao, Yuwei Zhao, Zhoukai Long, Saijuan Zhu, Junping Wu, Min Xiao, Jing Zhou, Shuo Zhang, Lianbin Li, and et al. 2023. "A Novel System for the Detection of Spontaneous Abortion-Causing Aneuploidy and Its Erroneous Chromosome Origins through the Combination of Low-Pass Copy Number Variation Sequencing and NGS-Based STR Tests" Journal of Clinical Medicine 12, no. 5: 1809. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12051809