
Elucidating the Molecular Pathways and Therapeutic Interventions of Gaseous Mediators in the Context of Fibrosis

Abstract
:1. Introduction
2. NO Either Suppresses or Promotes the Process of Fibrosis
2.1. NO Regulates ROS Production and HSCs’ Apoptosis in Liver Fibrosis
2.2. NO and NOS System in Pulmonary Fibrosis
2.3. NO Regulates ECM-Degrading Proteases in Renal Fibrosis

3. The Axis of HO/CO and Exogenous CO in Attenuation of Fibrosis
3.1. CO Inhibits the Proliferation and Activation of Fibroblasts in Pulmonary Fibrosis
3.2. CO Regulates MKK3, ERK-MAPK, and TGF-β/Smad Pathways in Renal Fibrosis
3.3. CO Modulates TGF-β Pathways and Autophagy in Myocardial Fibrosis
3.4. CO Attenuates Inflammation and Aging in Liver Fibrosis

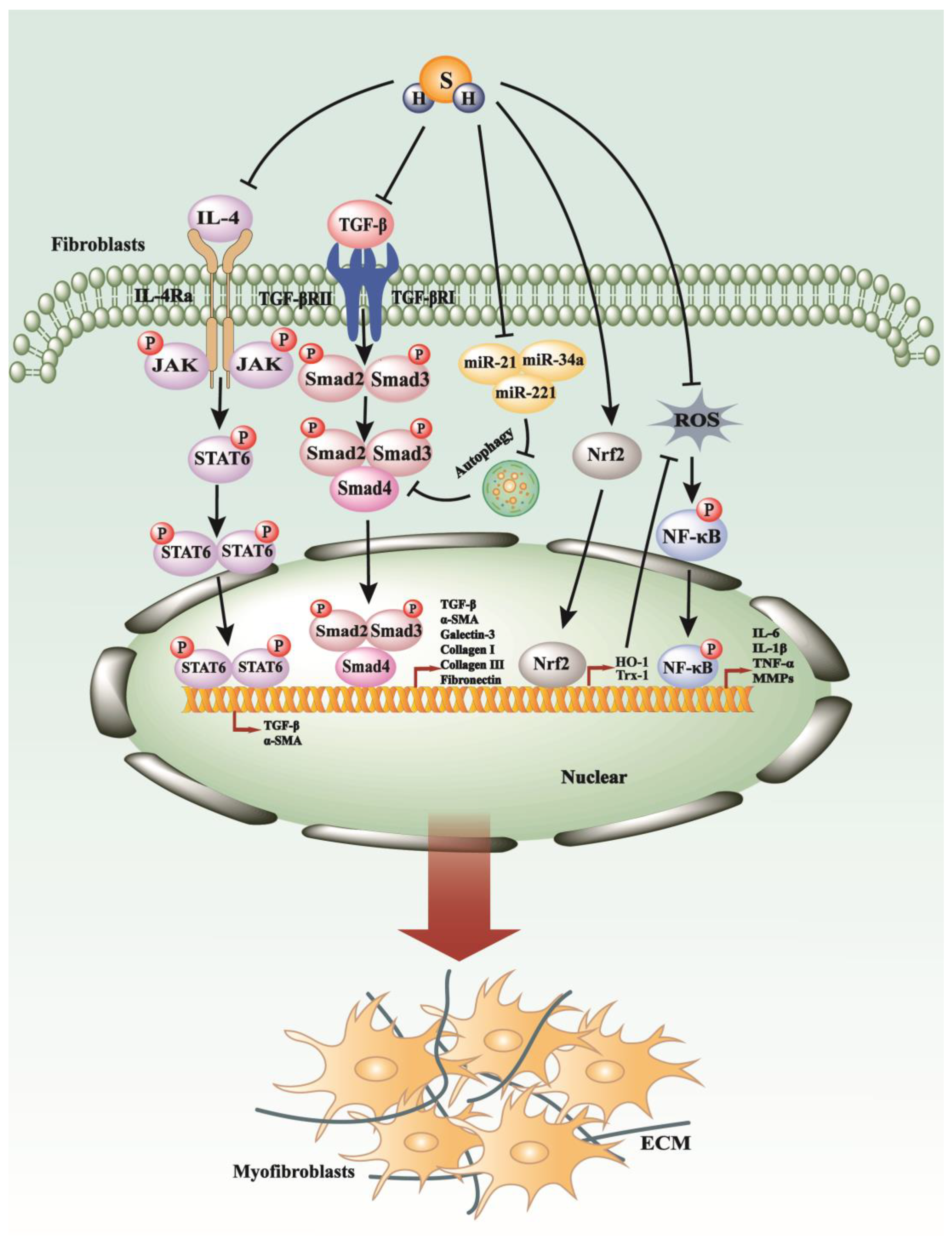
4. H2S Exerts Suppressive Impacts on the Process of Fibrosis
4.1. H2S Regulates TGF-β Pathways, Inflammation, Autophagy, and Oxidative Stress in Renal Fibrosis
4.2. H2S Modulates PI3K/AKT1, JAK/STAT, PKC-ERK1/2MAPK Pathways in Myocardial Fibrosis
4.3. H2S Regulates the Proliferation and Activation of HSCs in Liver Fibrosis

4.4. H2S Restrained the Inflammation and Oxidative Stress in Pulmonary Fibrosis
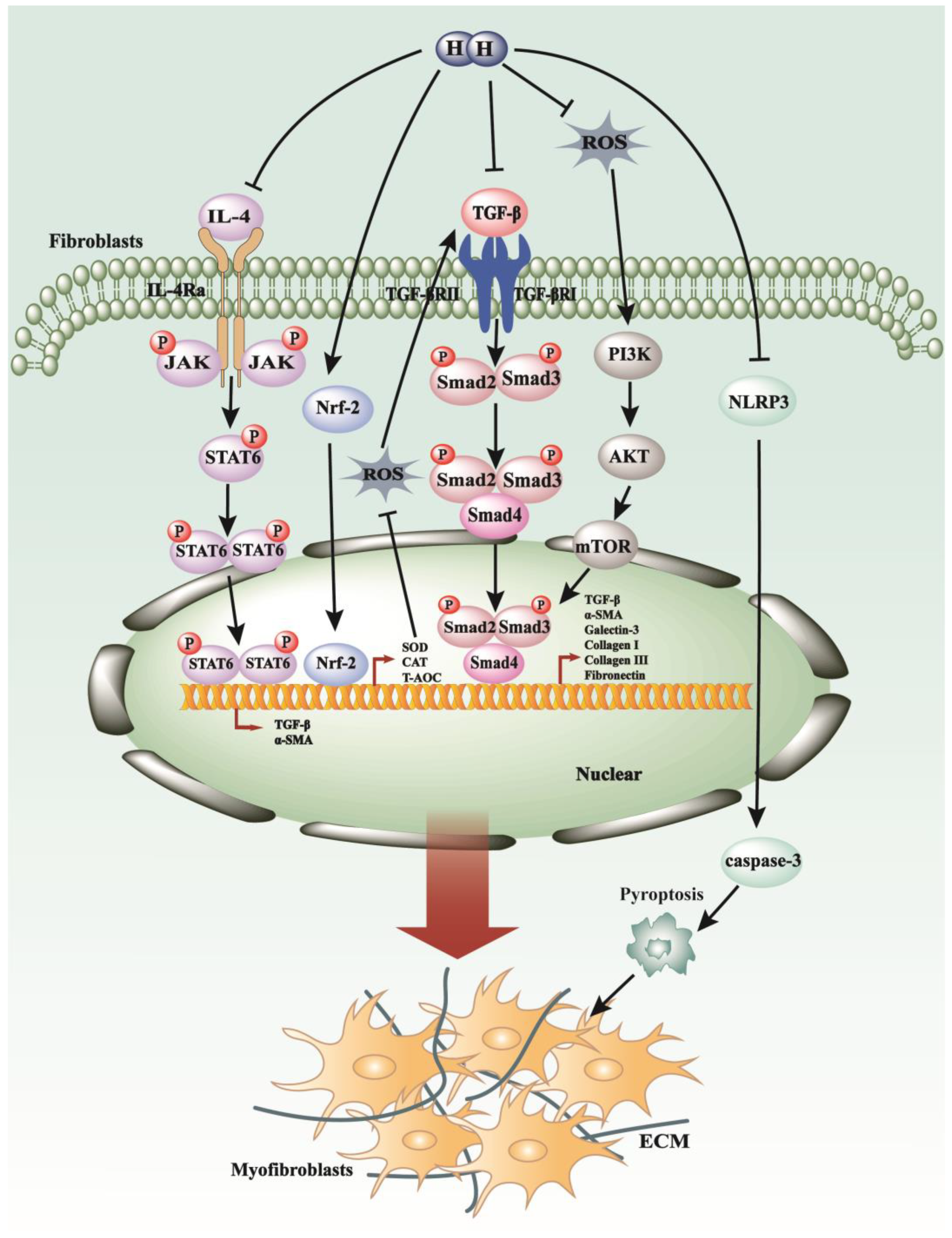
5. The Novel Gasotransmitter H2 in the Process of Fibrosis
5.1. H2 Regulates TGF-β1 Pathways, Oxidative Stress, and Inflammatory Response in Pulmonary Fibrosis
5.2. H2 Modulates the p38 MAPK, JAK/STAT Pathways, Pyroptosis, and Oxidative Stress in Myocardial Fibrosis
5.3. H2 Exerts a Suppressive Effect on the Development of Liver Fibrosis
5.4. H2 Regulates the Development of EMT and Oxidative Stress in Renal Fibrosis
5.5. H2 Plays a Suppressive Role in Peritoneal Fibrosis by Targeting TGF-β/PAI-1, Oxidative Stress, and Inflammation Pathways

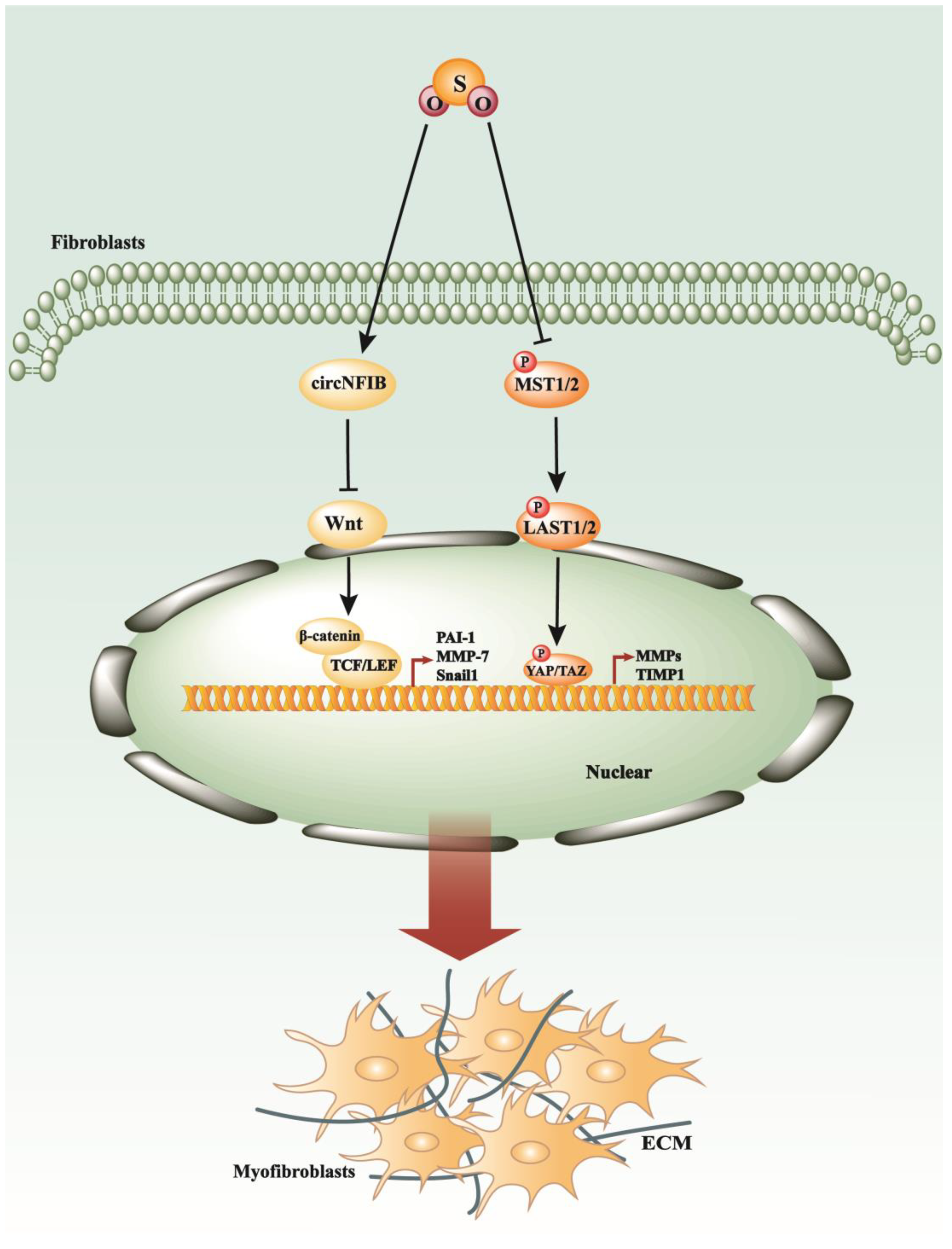
6. SO2 in the Process of Myocardial Fibrosis and Other Cardiovascular Diseases

7. Conclusions and Perspectives

Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wen, J.H.; Li, D.Y.; Liang, S.; Yang, C.; Tang, J.X.; Liu, H.F. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front. Immunol. 2022, 13, 946832. [Google Scholar] [CrossRef]
- Costa, C.; Sampaio-Maia, B.; Araujo, R.; Nascimento, D.S.; Ferreira-Gomes, J.; Pestana, M.; Azevedo, M.J.; Alencastre, I.S. Gut Microbiome and Organ Fibrosis. Nutrients 2022, 14, 352. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Krieg, T.; Abraham, D.; Lafyatis, R. Fibrosis in connective tissue disease: The role of the myofibroblast and fibroblast-epithelial cell interactions. Arthritis Res. Ther. 2007, 9 (Suppl. 2), S4. [Google Scholar] [CrossRef]
- Li, M.; Luan, F.; Zhao, Y.; Hao, H.; Zhou, Y.; Han, W.; Fu, X. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. 2016, 241, 1–13. [Google Scholar] [CrossRef]
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef]
- Trachtman, H.; Futterweit, S.; Singhal, P. Nitric oxide modulates the synthesis of extracellular matrix proteins in cultured rat mesangial cells. Biochem. Biophys. Res. Commun. 1995, 207, 120–125. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, F.; Kong, G.; Yuan, S.; Cao, Y.; Zhang, Q.; Wang, Q.; Liu, L. Gasotransmitter CO Attenuates Bleomycin-Induced Fibroblast Senescence via Induction of Stress Granule Formation. Oxid. Med. Cell. Longev. 2021, 2021, 9926284. [Google Scholar] [CrossRef]
- Fan, H.N.; Wang, H.J.; Yang-Dan, C.R.; Ren, L.; Wang, C.; Li, Y.F.; Deng, Y. Protective effects of hydrogen sulfide on oxidative stress and fibrosis in hepatic stellate cells. Mol. Med. Rep. 2013, 7, 247–253. [Google Scholar] [CrossRef]
- Xing, Z.; Pan, W.; Zhang, J.; Xu, X.; Zhang, X.; He, X.; Fan, M. Hydrogen Rich Water Attenuates Renal Injury and Fibrosis by Regulation Transforming Growth Factor-β Induced Sirt1. Biol. Pharm. Bull. 2017, 40, 610–615. [Google Scholar] [CrossRef]
- Ignarro, L.J. Physiology and pathophysiology of nitric oxide. Kidney Int. Suppl. 1996, 55, S2–S5. [Google Scholar]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298 Pt 2, 249–258. [Google Scholar] [CrossRef]
- Liu, J.; Hughes, T.E.; Sessa, W.C. The first 35 amino acids and fatty acylation sites determine the molecular targeting of endothelial nitric oxide synthase into the Golgi region of cells: A green fluorescent protein study. J. Cell Biol. 1997, 137, 1525–1535. [Google Scholar] [CrossRef]
- Vallance, P. Nitric oxide: Therapeutic opportunities. Fundam. Clin. Pharmacol. 2003, 17, 1–10. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Saccomanno, S.; van Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver 2001, 21, 1–12. [Google Scholar] [CrossRef]
- Langer, D.A.; Das, A.; Semela, D.; Kang-Decker, N.; Hendrickson, H.; Bronk, S.F.; Katusic, Z.S.; Gores, G.J.; Shah, V.H. Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology 2008, 47, 1983–1993. [Google Scholar] [CrossRef]
- Ali, G.; Mohsin, S.; Khan, M.; Nasir, G.A.; Shams, S.; Khan, S.N.; Riazuddin, S. Nitric oxide augments mesenchymal stem cell ability to repair liver fibrosis. J. Transl. Med. 2012, 10, 75. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Kim, M.Y. Nitric oxide in liver diseases. Trends Pharmacol. Sci. 2015, 36, 524–536. [Google Scholar] [CrossRef]
- Diesen, D.L.; Kuo, P.C. Nitric oxide and redox regulation in the liver: Part II. Redox biology in pathologic hepatocytes and implications for intervention. J. Surg. Res. 2011, 167, 96–112. [Google Scholar] [CrossRef]
- Anavi, S.; Eisenberg-Bord, M.; Hahn-Obercyger, M.; Genin, O.; Pines, M.; Tirosh, O. The role of iNOS in cholesterol-induced liver fibrosis. Lab. Investig. 2015, 95, 914–924. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, J.; Zhao, T.; Wang, L.; Wang, J. Modulation of the VEGF/AKT/eNOS signaling pathway to regulate liver angiogenesis to explore the anti-hepatic fibrosis mechanism of curcumol. J. Ethnopharmacol. 2021, 280, 114480. [Google Scholar] [CrossRef]
- Ali, F.E.M.; Azouz, A.A.; Bakr, A.G.; Abo-Youssef, A.M.; Hemeida, R.A.M. Hepatoprotective effects of diosmin and/or sildenafil against cholestatic liver cirrhosis: The role of Keap-1/Nrf-2 and P(38)-MAPK/NF-κB/iNOS signaling pathway. Food Chem. Toxicol. 2018, 120, 294–304. [Google Scholar] [CrossRef]
- Oltulu, F.; Buhur, A.; Gürel, Ç.; Kuşçu, G.C.; Dağdeviren, M.; Karabay Yavaşoğlu, N.; Köse, T.; Yavaşoğlu, A. Mid-dose losartan mitigates diabetes-induced hepatic damage by regulating iNOS, eNOS, VEGF, and NF-κB expressions. Turk. J. Med. Sci. 2019, 49, 1582–1589. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Noguchi, S.; Yatera, K.; Wang, K.Y.; Oda, K.; Akata, K.; Yamasaki, K.; Kawanami, T.; Ishimoto, H.; Toyohira, Y.; Shimokawa, H.; et al. Nitric oxide exerts protective effects against bleomycin-induced pulmonary fibrosis in mice. Respir. Res. 2014, 15, 92. [Google Scholar] [CrossRef]
- Cameli, P.; Bergantini, L.; Salvini, M.; Refini, R.M.; Pieroni, M.; Bargagli, E.; Sestini, P. Alveolar concentration of nitric oxide as a prognostic biomarker in idiopathic pulmonary fibrosis. Nitric Oxide 2019, 89, 41–45. [Google Scholar] [CrossRef]
- Theodoro-Júnior, O.A.; Righetti, R.F.; Almeida-Reis, R.; Martins-Oliveira, B.T.; Oliva, L.V.; Prado, C.M.; Saraiva-Romanholo, B.M.; Leick, E.A.; Pinheiro, N.M.; Lobo, Y.A.; et al. A Plant Proteinase Inhibitor from Enterolobium contortisiliquum Attenuates Pulmonary Mechanics, Inflammation and Remodeling Induced by Elastase in Mice. Int. J. Mol. Sci. 2017, 18, 403. [Google Scholar] [CrossRef]
- Saghir, S.A.M.; Al-Gabri, N.A.; Khafaga, A.F.; El-Shaer, N.H.; Alhumaidh, K.A.; Elsadek, M.F.; Ahmed, B.M.; Alkhawtani, D.M.; Abd El-Hack, M.E. Thymoquinone-PLGA-PVA Nanoparticles Ameliorate Bleomycin-Induced Pulmonary Fibrosis in Rats via Regulation of Inflammatory Cytokines and iNOS Signaling. Animals 2019, 9, 951. [Google Scholar] [CrossRef]
- Zaafan, M.A.; Haridy, A.R.; Abdelhamid, A.M. Amitriptyline attenuates bleomycin-induced pulmonary fibrosis: Modulation of the expression of NF-κβ, iNOS, and Nrf2. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 279–286. [Google Scholar] [CrossRef]
- Gungor, H.; Ekici, M.; Onder Karayigit, M.; Turgut, N.H.; Kara, H.; Arslanbas, E. Zingerone ameliorates oxidative stress and inflammation in bleomycin-induced pulmonary fibrosis: Modulation of the expression of TGF-β1 and iNOS. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1659–1670. [Google Scholar] [CrossRef]
- Nathan, S.D.; Flaherty, K.R.; Glassberg, M.K.; Raghu, G.; Swigris, J.; Alvarez, R.; Ettinger, N.; Loyd, J.; Fernandes, P.; Gillies, H.; et al. A Randomized, Double-Blind, Placebo-Controlled Study of Pulsed, Inhaled Nitric Oxide in Subjects at Risk of Pulmonary Hypertension Associated with Pulmonary Fibrosis. Chest 2020, 158, 637–645. [Google Scholar] [CrossRef]
- Klinkhammer, B.M.; Goldschmeding, R.; Floege, J.; Boor, P. Treatment of Renal Fibrosis-Turning Challenges into Opportunities. Adv. Chronic Kidney Dis. 2017, 24, 117–129. [Google Scholar] [CrossRef]
- Dolman, M.E.; Harmsen, S.; Storm, G.; Hennink, W.E.; Kok, R.J. Drug targeting to the kidney: Advances in the active targeting of therapeutics to proximal tubular cells. Adv. Drug Deliv. Rev. 2010, 62, 1344–1357. [Google Scholar] [CrossRef]
- Nastase, M.V.; Zeng-Brouwers, J.; Wygrecka, M.; Schaefer, L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Adv. Drug Deliv. Rev. 2018, 129, 295–307. [Google Scholar] [CrossRef]
- Wani, J.; Carl, M.; Henger, A.; Nelson, P.J.; Rupprecht, H. Nitric oxide modulates expression of extracellular matrix genes linked to fibrosis in kidney mesangial cells. Biol. Chem. 2007, 388, 497–506. [Google Scholar] [CrossRef]
- Chen, Y.; Yuan, S.; Cao, Y.; Kong, G.; Jiang, F.; Li, Y.; Wang, Q.; Tang, M.; Zhang, Q.; Wang, Q.; et al. Gasotransmitters: Potential Therapeutic Molecules of Fibrotic Diseases. Oxid. Med. Cell. Longev. 2021, 2021, 3206982. [Google Scholar] [CrossRef]
- Murrell, G.A.; Jang, D.; Williams, R.J. Nitric oxide activates metalloprotease enzymes in articular cartilage. Biochem. Biophys. Res. Commun. 1995, 206, 15–21. [Google Scholar] [CrossRef]
- Woessner, J.F., Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar] [CrossRef]
- Eberhardt, W.; Beeg, T.; Beck, K.F.; Walpen, S.; Gauer, S.; Böhles, H.; Pfeilschifter, J. Nitric oxide modulates expression of matrix metalloproteinase-9 in rat mesangial cells. Kidney Int. 2000, 57, 59–69. [Google Scholar] [CrossRef]
- Eberhardt, W.; Beck, K.F.; Pfeilschifter, J. Cytokine-induced expression of tPA is differentially modulated by NO and ROS in rat mesangial cells. Kidney Int. 2002, 61, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Akool, E.S.; Doller, A.; Müller, R.; Gutwein, P.; Xin, C.; Huwiler, A.; Pfeilschifter, J.; Eberhardt, W. Nitric oxide induces TIMP-1 expression by activating the transforming growth factor beta-Smad signaling pathway. J. Biol. Chem. 2005, 280, 39403–39416. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, Y.; Liu, C.; Zhou, X.; Li, X.; Xiao, A. Effects of nitric oxide on renal interstitial fibrosis in rats with unilateral ureteral obstruction. Life Sci. 2012, 90, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Lu, H.H.; Cheng, H.T.; Huang, H.C.; Tsai, Y.J.; Chang, I.H.; Tu, C.P.; Chung, C.W.; Lu, T.T.; Peng, C.H.; et al. Delivery of nitric oxide with a pH-responsive nanocarrier for the treatment of renal fibrosis. J. Control. Release 2023, 354, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.J.; Wang, L.; Xu, Q.; McTiernan, C.F.; Shiva, S.; Tejero, J.; Gladwin, M.T. Carbon Monoxide Poisoning: Pathogenesis, Management, and Future Directions of Therapy. Am. J. Respir. Crit. Care Med. 2017, 195, 596–606. [Google Scholar] [CrossRef]
- Nagao, S.; Taguchi, K.; Sakai, H.; Tanaka, R.; Horinouchi, H.; Watanabe, H.; Kobayashi, K.; Otagiri, M.; Maruyama, T. Carbon monoxide-bound hemoglobin-vesicles for the treatment of bleomycin-induced pulmonary fibrosis. Biomaterials 2014, 35, 6553–6562. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, G.; Yoshida, T.; Noguchi, M. Heme oxygenase and heme degradation. Biochem. Biophys Res. Commun. 2005, 338, 558–567. [Google Scholar] [CrossRef]
- Montellano, P.R. The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 2000, 4, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Foresti, R.; Motterlini, R. The heme oxygenase pathway and its interaction with nitric oxide in the control of cellular homeostasis. Free Radic. Res. 1999, 31, 459–475. [Google Scholar] [CrossRef]
- Motterlini, R.; Mann, B.E.; Foresti, R. Therapeutic applications of carbon monoxide-releasing molecules. Expert Opin. Investig. Drugs 2005, 14, 1305–1318. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Pereira, C.; Dias-Pedroso, D.; Soares, N.L.; Vieira, H.L.A. CO-mediated cytoprotection is dependent on cell metabolism modulation. Redox. Biol. 2020, 32, 101470. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Zeb, A.; Kim, M.S.; Rana, I.; Khan, N.; Qureshi, O.S.; Lim, C.W.; Park, J.S.; Gao, Z.; Maeng, H.J.; et al. Controlled therapeutic delivery of CO from carbon monoxide-releasing molecules (CORMs). J. Control. Release 2022, 350, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Adach, W.; Błaszczyk, M.; Olas, B. Carbon monoxide and its donors—Chemical and biological properties. Chem. Biol. Interact. 2020, 318, 108973. [Google Scholar] [CrossRef] [PubMed]
- Foresti, R.; Bani-Hani, M.G.; Motterlini, R. Use of carbon monoxide as a therapeutic agent: Promises and challenges. Intensive Care Med. 2008, 34, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R. Carbon monoxide-releasing molecules (CO-RMs): Vasodilatory, anti-ischaemic and anti-inflammatory activities. Biochem. Soc. Trans. 2007, 35, 1142–1146. [Google Scholar] [CrossRef]
- Zhou, Z.; Song, R.; Fattman, C.L.; Greenhill, S.; Alber, S.; Oury, T.D.; Choi, A.M.; Morse, D. Carbon monoxide suppresses bleomycin-induced lung fibrosis. Am. J. Pathol. 2005, 166, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Li, J.C.; Wang, S.M.; Cheng, C.H.; Yeh, C.H.; Lin, L.S.; Chiu, H.Y.; Chang, C.Y.; Chuu, J.J. The effects of carbon monoxide releasing molecules on paraquat-induced pulmonary interstitial inflammation and fibrosis. Toxicology 2021, 456, 152750. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Rosas, I.O.; Goldberg, H.J.; Collard, H.R.; El-Chemaly, S.; Flaherty, K.; Hunninghake, G.M.; Lasky, J.A.; Lederer, D.J.; Machado, R.; Martinez, F.J.; et al. A Phase II Clinical Trial of Low-Dose Inhaled Carbon Monoxide in Idiopathic Pulmonary Fibrosis. Chest 2018, 153, 94–104. [Google Scholar] [CrossRef]
- Neto, J.S.; Nakao, A.; Toyokawa, H.; Nalesnik, M.A.; Romanosky, A.J.; Kimizuka, K.; Kaizu, T.; Hashimoto, N.; Azhipa, O.; Stolz, D.B.; et al. Low-dose carbon monoxide inhalation prevents development of chronic allograft nephropathy. Am. J. Physiol. Renal Physiol. 2006, 290, F324–F334. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lee, J.Y.; Kwak, J.H.; He, Y.; Kim, S.I.; Choi, M.E. Protective effects of low-dose carbon monoxide against renal fibrosis induced by unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 2008, 294, F508–F517. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Faleo, G.; Nalesnik, M.A.; Seda-Neto, J.; Kohmoto, J.; Murase, N. Low-dose carbon monoxide inhibits progressive chronic allograft nephropathy and restores renal allograft function. Am. J. Physiol. Renal Physiol. 2009, 297, F19–F26. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.; Elhadad, S.; Robison, T.; Terry, H.; Varshney, R.; Woolington, S.; Ghafoory, S.; Choi, M.E.; Ahamed, J. HIV protease inhibitor-induced cardiac dysfunction and fibrosis is mediated by platelet-derived TGF-β1 and can be suppressed by exogenous carbon monoxide. PLoS ONE 2017, 12, e0187185. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Guo, C.; Xia, Z.; Xue, Y.; Song, B.; Hu, W.; He, X.; Liang, S.; Wei, Y.; Zhang, C.; et al. Exploring the therapeutic potential of a nano micelle containing a carbon monoxide-releasing molecule for metabolic-associated fatty liver disease by modulating hypoxia-inducible factor-1α. Acta Biomater. 2023, 169, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Zhang, H.M.; Li, J.X.; Li, Y.; Wang, Q.; Kong, G.Y.; Li, A.H.; Nan, J.X.; Chen, Y.Q.; Zhang, Q.G. Gasotransmitters in non-alcoholic fatty liver disease: Just the tip of the iceberg. Eur. J. Pharmacol. 2023, 954, 175834. [Google Scholar] [CrossRef] [PubMed]
- Joe, Y.; Chen, Y.; Park, J.; Kim, H.J.; Rah, S.Y.; Ryu, J.; Cho, G.J.; Choi, H.S.; Ryter, S.W.; Park, J.W.; et al. Cross-talk between CD38 and TTP Is Essential for Resolution of Inflammation during Microbial Sepsis. Cell Rep. 2020, 30, 1063–1076.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Park, H.J.; Park, J.; Song, H.C.; Ryter, S.W.; Surh, Y.J.; Kim, U.H.; Joe, Y.; Chung, H.T. Carbon monoxide ameliorates acetaminophen-induced liver injury by increasing hepatic HO-1 and Parkin expression. FASEB J. 2019, 33, 13905–13919. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Joe, Y.; Park, J.; Song, H.C.; Kim, U.H.; Chung, H.T. Carbon monoxide induces the assembly of stress granule through the integrated stress response. Biochem. Biophys. Res. Commun. 2019, 512, 289–294. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Kim, J.; Hwang, E.; Park, G.H.; Yang, C.H.; Ryter, S.W.; Park, J.W.; Chung, H.T.; Joe, Y. CO-Induced TTP Activation Alleviates Cellular Senescence and Age-Dependent Hepatic Steatosis via Downregulation of PAI-1. Aging Dis. 2023, 14, 484–501. [Google Scholar] [CrossRef]
- Zaorska, E.; Tomasova, L.; Koszelewski, D.; Ostaszewski, R.; Ufnal, M. Hydrogen Sulfide in Pharmacotherapy, Beyond the Hydrogen Sulfide-Donors. Biomolecules 2020, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Shibuya, N.; Kimura, Y.; Nagahara, N.; Ogasawara, Y.; Kimura, H. Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 2011, 439, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.L.; Li, N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Pan, C.; Zhou, F.; Yuan, Z.; Wang, H.; Cui, W.; Zhang, G. Hydrogen Sulfide as a Potential Therapeutic Target in Fibrosis. Oxid. Med. Cell. Longev. 2015, 2015, 593407. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Dugbartey, G.J.; Talaei, F.; Houwertjes, M.C.; Goris, M.; Epema, A.H.; Bouma, H.R.; Henning, R.H. Dopamine treatment attenuates acute kidney injury in a rat model of deep hypothermia and rewarming—The role of renal H2S-producing enzymes. Eur. J. Pharmacol. 2015, 769, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Dugbartey, G.J.; Bouma, H.R.; Strijkstra, A.M.; Boerema, A.S.; Henning, R.H. Induction of a Torpor-Like State by 5′-AMP Does Not Depend on H2S Production. PLoS ONE 2015, 10, e0136113. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, J.; Xie, F.; Tan, W.; Wang, S.; Huang, L.; Tao, L.; Xing, Q.; Yuan, Q. Loss of the Protein Cystathionine β-Synthase During Kidney Injury Promotes Renal Tubulointerstitial Fibrosis. Kidney Blood Press. Res. 2017, 42, 428–443. [Google Scholar] [CrossRef]
- Song, K.; Wang, F.; Li, Q.; Shi, Y.B.; Zheng, H.F.; Peng, H.; Shen, H.Y.; Liu, C.F.; Hu, L.F. Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int. 2014, 85, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine γ-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.J.; Jang, H.S.; Kim, J.I.; Han, S.J.; Park, J.W.; Park, K.M. Involvement of hydrogen sulfide and homocysteine transsulfuration pathway in the progression of kidney fibrosis after ureteral obstruction. Biochim. Biophys. Acta 2013, 1832, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, X.; Wang, X.; Peng, Y.; Du, J.; Yin, H.; Yang, H.; Ni, X.; Zhang, W. H2S alleviates renal injury and fibrosis in response to unilateral ureteral obstruction by regulating macrophage infiltration via inhibition of NLRP3 signaling. Exp. Cell Res. 2020, 387, 111779. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, J.; Zhang, H.; Shen, Z.; Liu, H.; Lv, S.; Yu, X.; Zhang, D.; Ding, X.; Zhang, X. Hydrogen sulfide attenuates renal fibrosis by inducing TET-dependent DNA demethylation on Klotho promoter. FASEB J. 2020, 34, 11474–11487. [Google Scholar] [CrossRef] [PubMed]
- Zeng, O.; Li, F.; Li, Y.; Li, L.; Xiao, T.; Chu, C.; Yang, J. Effect of Novel Gasotransmitter hydrogen sulfide on renal fibrosis and connexins expression in diabetic rats. Bioengineered 2016, 7, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xiao, T.; Li, F.; Li, Y.; Zeng, O.; Liu, M.; Liang, B.; Li, Z.; Chu, C.; Yang, J. Hydrogen sulfide reduced renal tissue fibrosis by regulating autophagy in diabetic rats. Mol. Med. Rep. 2017, 16, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, L.; Zeng, O.; Liu, J.M.; Yang, J. H2S improves renal fibrosis in STZ-induced diabetic rats by ameliorating TGF-β1 expression. Ren. Fail. 2017, 39, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Snijder, P.M.; Frenay, A.R.; de Boer, R.A.; Pasch, A.; Hillebrands, J.L.; Leuvenink, H.G.; van Goor, H. Exogenous administration of thiosulfate, a donor of hydrogen sulfide, attenuates angiotensin II-induced hypertensive heart disease in rats. Br. J. Pharmacol. 2015, 172, 1494–1504. [Google Scholar] [CrossRef]
- Lilyanna, S.; Peh, M.T.; Liew, O.W.; Wang, P.; Moore, P.K.; Richards, A.M.; Martinez, E.C. GYY4137 attenuates remodeling, preserves cardiac function and modulates the natriuretic peptide response to ischemia. J. Mol. Cell. Cardiol. 2015, 87, 27–37. [Google Scholar] [CrossRef]
- Pan, L.L.; Wang, X.L.; Wang, X.L.; Zhu, Y.Z. Sodium hydrosulfide prevents myocardial dysfunction through modulation of extracellular matrix accumulation and vascular density. Int. J. Mol. Sci. 2014, 15, 23212–23226. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, D.; Zheng, J.; Huang, X.; Jin, H. Hydrogen sulfide attenuates cardiac hypertrophy and fibrosis induced by abdominal aortic coarctation in rats. Mol. Med. Rep. 2012, 5, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zeng, O.; Luo, J.; Wu, Z.; Li, F.; Yang, J. Effects of hydrogen sulfide on myocardial fibrosis in diabetic rats: Changes in matrix metalloproteinases parameters. Biomed. Mater. Eng. 2015, 26 (Suppl. 1), S2033–S2039. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhao, L.; Mao, J.; Huang, J.; Chen, J. Antioxidant effects of hydrogen sulfide on left ventricular remodeling in smoking rats are mediated via PI3K/Akt-dependent activation of Nrf2. Toxicol. Sci. 2015, 144, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Luo, J.; Wu, Z.; Li, F.; Zeng, O.; Yang, J. Effects of hydrogen sulfide on myocardial fibrosis and PI3K/AKT1-regulated autophagy in diabetic rats. Mol. Med. Rep. 2016, 13, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Xiao, T.; Long, J.; Liu, M.; Li, Z.; Liu, S.; Yang, J. Hydrogen sulfide alleviates myocardial fibrosis in mice with alcoholic cardiomyopathy by downregulating autophagy. Int. J. Mol. Med. 2017, 40, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Y.; Liang, B.; Li, Z.; Jiang, Z.; Chu, C.; Yang, J. Hydrogen sulfide attenuates myocardial fibrosis in diabetic rats through the JAK/STAT signaling pathway. Int. J. Mol. Med. 2018, 41, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Z.; Liang, B.; Li, L.; Liu, S.; Tan, W.; Long, J.; Tang, F.; Chu, C.; Yang, J. Hydrogen sulfide ameliorates rat myocardial fibrosis induced by thyroxine through PI3K/AKT signaling pathway. Endocr. J. 2018, 65, 769–781. [Google Scholar] [CrossRef]
- Long, J.; Liu, M.; Liu, S.; Tang, F.; Tan, W.; Xiao, T.; Chu, C.; Yang, J. H2S attenuates the myocardial fibrosis in diabetic rats through modulating PKC-ERK1/2MAPK signaling pathway. Technol. Health Care 2019, 27, 307–316. [Google Scholar] [CrossRef]
- Yang, R.; Jia, Q.; Ma, S.F.; Wang, Y.; Mehmood, S.; Chen, Y. Exogenous H2S mitigates myocardial fibrosis in diabetic rats through suppression of the canonical Wnt pathway. Int. J. Mol. Med. 2019, 44, 549–558. [Google Scholar] [CrossRef]
- Nie, L.; Liu, M.; Chen, J.; Wu, Q.; Li, Y.; Yi, J.; Zheng, X.; Zhang, J.; Chu, C.; Yang, J. Hydrogen sulfide ameliorates doxorubicin-induced myocardial fibrosis in rats via the PI3K/AKT/mTOR pathway. Mol. Med. Rep. 2021, 23, 299. [Google Scholar] [CrossRef]
- Su, H.; Su, H.; Liu, C.H.; Hu, H.J.; Zhao, J.B.; Zou, T.; Tang, Y.X. H2S inhibits atrial fibrillation-induced atrial fibrosis through miR-133a/CTGF axis. Cytokine 2021, 146, 155557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gong, W.; Xu, M.; Zhang, S.; Shen, J.; Zhu, M.; Wang, Y.; Chen, Y.; Shi, J.; Meng, G. Necroptosis Inhibition by Hydrogen Sulfide Alleviated Hypoxia-Induced Cardiac Fibroblasts Proliferation via Sirtuin 3. Int. J. Mol. Sci. 2021, 22, 11893. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Mencarelli, A.; Orlandi, S.; Renga, B.; Rizzo, G.; Distrutti, E.; Shah, V.; Morelli, A. The third gas: H2S regulates perfusion pressure in both the isolated and perfused normal rat liver and in cirrhosis. Hepatology 2005, 42, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Pan, S.; Li, J.; Dong, X.; Kang, K.; Zhao, M.; Jiang, X.; Kanwar, J.R.; Qiao, H.; Jiang, H.; et al. Hydrogen sulfide attenuates carbon tetrachloride-induced hepatotoxicity, liver cirrhosis and portal hypertension in rats. PLoS ONE 2011, 6, e25943. [Google Scholar] [CrossRef]
- Fan, H.N.; Chen, N.W.; Shen, W.L.; Zhao, X.Y.; Zhang, J. Endogenous hydrogen sulfide is associated with angiotensin II type 1 receptor in a rat model of carbon tetrachloride-induced hepatic fibrosis. Mol. Med. Rep. 2015, 12, 3351–3358. [Google Scholar] [CrossRef]
- Damba, T.; Zhang, M.; Buist-Homan, M.; van Goor, H.; Faber, K.N.; Moshage, H. Hydrogen sulfide stimulates activation of hepatic stellate cells through increased cellular bio-energetics. Nitric Oxide 2019, 92, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef]
- Fang, L.; Li, H.; Tang, C.; Geng, B.; Qi, Y.; Liu, X. Hydrogen sulfide attenuates the pathogenesis of pulmonary fibrosis induced by bleomycin in rats. Can. J. Physiol. Pharmacol. 2009, 87, 531–538. [Google Scholar] [CrossRef]
- Fang, L.P.; Lin, Q.; Tang, C.S.; Liu, X.M. Hydrogen sulfide attenuates epithelial-mesenchymal transition of human alveolar epithelial cells. Pharmacol. Res. 2010, 61, 298–305. [Google Scholar] [CrossRef]
- Cao, H.; Zhou, X.; Zhang, J.; Huang, X.; Zhai, Y.; Zhang, X.; Chu, L. Hydrogen sulfide protects against bleomycin-induced pulmonary fibrosis in rats by inhibiting NF-κB expression and regulating Th1/Th2 balance. Toxicol. Lett. 2014, 224, 387–394. [Google Scholar] [CrossRef]
- Zhou, X.; An, G.; Chen, J. Inhibitory effects of hydrogen sulphide on pulmonary fibrosis in smoking rats via attenuation of oxidative stress and inflammation. J. Cell. Mol. Med. 2014, 18, 1098–1103. [Google Scholar] [CrossRef]
- Dole, M.; Wilson, F.R.; Fife, W.P. Hyperbaric hydrogen therapy: A possible treatment for cancer. Science 1975, 190, 152–154. [Google Scholar] [CrossRef]
- Fu, Z.; Zhang, J. Molecular hydrogen is a promising therapeutic agent for pulmonary disease. J. Zhejiang Univ. Sci. B 2022, 23, 102–122. [Google Scholar] [CrossRef]
- Sun, H.; Chen, L.; Zhou, W.; Hu, L.; Li, L.; Tu, Q.; Chang, Y.; Liu, Q.; Sun, X.; Wu, M.; et al. The protective role of hydrogen-rich saline in experimental liver injury in mice. J. Hepatol. 2011, 54, 471–480. [Google Scholar] [CrossRef]
- Dong, W.W.; Zhang, Y.Q.; Zhu, X.Y.; Mao, Y.F.; Sun, X.J.; Liu, Y.J.; Jiang, L. Protective Effects of Hydrogen-Rich Saline Against Lipopolysaccharide-Induced Alveolar Epithelial-to-Mesenchymal Transition and Pulmonary Fibrosis. Med. Sci. Monit. 2017, 23, 2357–2364. [Google Scholar] [CrossRef]
- Li, T.; Deng, S.; Lei, W.; Li, Z.; Wu, W.; Zhang, T.; Dong, Z. Hydrogen water alleviates paraquat-induced lung fibroblast injury in vitro by enhancing Nrf2 expression. Nan Fang Yi Ke Da Xue Xue Bao 2020, 40, 233–239. [Google Scholar] [CrossRef]
- Gao, L.; Jiang, D.; Geng, J.; Dong, R.; Dai, H. Hydrogen inhalation attenuated bleomycin-induced pulmonary fibrosis by inhibiting transforming growth factor-β1 and relevant oxidative stress and epithelial-to-mesenchymal transition. Exp. Physiol. 2019, 104, 1942–1951. [Google Scholar] [CrossRef]
- Aokage, T.; Seya, M.; Hirayama, T.; Nojima, T.; Iketani, M.; Ishikawa, M.; Terasaki, Y.; Taniguchi, A.; Miyahara, N.; Nakao, A.; et al. The effects of inhaling hydrogen gas on macrophage polarization, fibrosis, and lung function in mice with bleomycin-induced lung injury. BMC Pulm. Med. 2021, 21, 339. [Google Scholar] [CrossRef]
- Braunwald, E. The war against heart failure: The Lancet lecture. Lancet 2015, 385, 812–824. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, S.; Zhu, L.; Cai, J.; Fu, L. Hydrogen-containing saline alleviates pressure overload-induced interstitial fibrosis and cardiac dysfunction in rats. Mol. Med. Rep. 2017, 16, 1771–1778. [Google Scholar] [CrossRef]
- Fan, Z.; Gao, Y.; Huang, Z.; Xue, F.; Wu, S.; Yang, J.; Zhu, L.; Fu, L. Protective effect of hydrogen-rich saline on pressure overload-induced cardiac hypertrophyin rats: Possible role of JAK-STAT signaling. BMC Cardiovasc. Disord. 2018, 18, 32. [Google Scholar] [CrossRef]
- Nie, C.; Zou, R.; Pan, S.; A, R.; Gao, Y.; Yang, H.; Bai, J.; Xi, S.; Wang, X.; Hong, X. Hydrogen gas inhalation ameliorates cardiac remodelling and fibrosis by regulating NLRP3 inflammasome in myocardial infarction rats. J. Cell. Mol. Med. 2021, 25, 8997–9010. [Google Scholar] [CrossRef]
- Liu, L.; Shi, Q.; Liu, X.; Li, Y.; Li, X. Attenuation of Myocardial Fibrosis Using Molecular Hydrogen by Inhibiting the TGF-β Signaling Pathway in Spontaneous Hypertensive Rats. Am. J. Hypertens. 2022, 35, 156–163. [Google Scholar] [CrossRef]
- Zhai, X.; Chen, X.; Lu, J.; Zhang, Y.; Sun, X.; Huang, Q.; Wang, Q. Hydrogen-rich saline improves non-alcoholic fatty liver disease by alleviating oxidative stress and activating hepatic PPARα and PPARγ. Mol. Med. Rep. 2017, 15, 1305–1312. [Google Scholar] [CrossRef]
- Li, S.W.; Takahara, T.; Que, W.; Fujino, M.; Guo, W.Z.; Hirano, S.I.; Ye, L.P.; Li, X.K. Hydrogen-rich water protects against liver injury in nonalcoholic steatohepatitis through HO-1 enhancement via IL-10 and Sirt 1 signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G450–G463. [Google Scholar] [CrossRef]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Y.B.; Li, Z.Z.; Yang, M.W.; Wang, S.; Jiang, D.P. Hydrogen-rich saline ameliorates renal injury induced by unilateral ureteral obstruction in rats. Int. Immunopharmacol. 2013, 17, 447–452. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, S.; Gu, C.; Pei, H.; Hong, X. Effect of Hydrogen-Rich Saline on Postoperative Intra-Abdominal Adhesion Bands Formation in Mice. Med. Sci. Monit. 2017, 23, 5363–5373. [Google Scholar] [CrossRef]
- Mehrotra, R.; Devuyst, O.; Davies, S.J.; Johnson, D.W. The Current State of Peritoneal Dialysis. J. Am. Soc. Nephrol. 2016, 27, 3238–3252. [Google Scholar] [CrossRef]
- Lu, H.; Chen, W.; Liu, W.; Si, Y.; Zhao, T.; Lai, X.; Kang, Z.; Sun, X.; Guo, Z. Molecular hydrogen regulates PTEN-AKT-mTOR signaling via ROS to alleviate peritoneal dialysis-related peritoneal fibrosis. FASEB J. 2020, 34, 4134–4146. [Google Scholar] [CrossRef]
- Cartledge, B.T.; Marcotte, A.R.; Herckes, P.; Anbar, A.D.; Majestic, B.J. The impact of particle size, relative humidity, and sulfur dioxide on iron solubility in simulated atmospheric marine aerosols. Environ. Sci. Technol. 2015, 49, 7179–7187. [Google Scholar] [CrossRef]
- Liu, M.; Liu, S.; Tan, W.; Tang, F.; Long, J.; Li, Z.; Liang, B.; Chu, C.; Yang, J. Gaseous signalling molecule SO2 via Hippo-MST pathway to improve myocardial fibrosis of diabetic rats. Mol. Med. Rep. 2017, 16, 8953–8963. [Google Scholar] [CrossRef]
- Cai, H.Y.; Li, B.; Dang, L.; Yang, J.; Man, K.; Dong, C.M.; Lu, Y. Sulfur dioxide in the caudal ventrolateral medulla reduces blood pressure and heart rate in rats via the glutamate receptor and NOS/cGMP signal pathways. Sheng Li Xue Bao 2023, 75, 27–35. [Google Scholar]
- Huang, Y.; Tang, C.; Du, J.; Jin, H. Endogenous Sulfur Dioxide: A New Member of Gasotransmitter Family in the Cardiovascular System. Oxidative Med. Cell. Longev. 2016, 2016, 8961951. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, R.; Wang, D.; Lin, Y.; Bai, C.; Nie, N.; Gao, S.; Zhang, Q.; Chang, H.; Ren, C. Elucidating the role of circNFIB in myocardial fibrosis alleviation by endogenous sulfur dioxide. BMC Cardiovasc. Disord. 2022, 22, 492. [Google Scholar] [CrossRef]
- Zhang, L.L.; Du, J.B.; Tang, C.S.; Jin, H.F.; Huang, Y.Q. Inhibitory Effects of Sulfur Dioxide on Rat Myocardial Fibroblast Proliferation and Migration. Chin. Med. J. 2018, 131, 1715–1723. [Google Scholar] [CrossRef]
- Zhang, S.; Qiu, B.; Lv, B.; Yang, G.; Tao, Y.; Hu, Y.; Li, K.; Yu, X.; Tang, C.; Du, J.; et al. Endogenous sulfur dioxide deficiency as a driver of cardiomyocyte senescence through abolishing sulphenylation of STAT3 at cysteine 259. Redox Biol. 2024, 71, 103124. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, L.; Chen, S.; Huang, Y.; Li, K.; Yu, X.; Wu, H.; Tian, X.; Zhang, C.; Tang, C.; et al. Downregulated endogenous sulfur dioxide/aspartate aminotransferase pathway is involved in angiotensin II-stimulated cardiomyocyte autophagy and myocardial hypertrophy in mice. Int. J. Cardiol. 2016, 225, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Du, J.; Jin, H.; Li, W.; Liang, Y.; Geng, B.; Li, S.; Zhang, C.; Tang, C. Endogenous sulfur dioxide aggravates myocardial injury in isolated rat heart with ischemia and reperfusion. Transplantation 2009, 87, 517–524. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, D.; Ochs, T.; Tang, C.; Chen, S.; Zhang, S.; Geng, B.; Jin, H.; Du, J. Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats. Lab. Investig. 2011, 91, 12–23. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gasotransmitters | Administration | Diseases | Mechanisms | Quotation |
|---|---|---|---|---|
| NO | SNAP | Liver fibrosis | SNAP eliminates ROS production, inhibits the production of ECM. | [15] |
| Liver fibrosis | SNAP donors promote HSC apoptosis and maintains sinus homeostasis by producing superoxide and hydroxyl radical intermediates. | [16] | ||
| Renal fibrosis | SNAP inhibits the proliferation of MCs, and down-regulates the expression of ECM. | [36] | ||
| Renal fibrosis | SNAP reduces fibrosis formation by inhibiting ECM synthesis. | [7,38,39] | ||
| Renal fibrosis | SNAP alleviates fibrosis by regulating the expression of several ECM-degrading proteases and intrinsic inhibitors in renal MCs, including MMP-9, MMP-13, plasminogen activator inhibitor-1 (PAI-1), and TIMP-1. | [40,41] | ||
| Renal fibrosis | SNAP exerts antifibrotic effects by amplifying TIMP-1 expression in renal MCs in a TGF-β-dependent manner. | [42] | ||
| SNP | Liver fibrosis | SNP inhibits CCl4 induced liver fibrosis via enhancing MSC. | [17] | |
| CO | CO gas | Idiopathic pulmonary fibrosis | CO gas upregulates the expression of p21 Cip1 to inhibit the proliferation of fibroblasts. | [57] |
| Renal fibrosis | 250 ppm CO activates MKK3 signaling pathway to inhibit the development of renal fibrosis and protect the kidney. | [62] | ||
| Renal fibrosis | CO suppresses the expression of TGF-β1/Smads and ERK-MAPK pathways to inhibit progression of fibroinflammatory process of CAN. | [63] | ||
| Myocardial fibrosis | CO inhibits TGF-β signaling and stimulating autophagy. | [64] | ||
| CORM-A1 | Idiopathic pulmonary fibrosis | CORM-A1 reduces the expression of profibrogenic cytokines COX-2, TNF-α, and α-SMA and serum hydroxyproline to alleviate paraquat-induced pulmonary fibrosis. | [58] | |
| SMA/CORM2 | Liver fibrosis | SMA/CORM2 inhibits HIF-1α-mediated inflammatory cascade. | [66] | |
| CORM2 | Liver fibrosis | CORM2 up-regulates TTP to reduce the level of PAI-1 in NAFLD. | [70] | |
| CO-HbV | Idiopathic pulmonary fibrosis | CO-HbV attenuates IPF through suppressing NOX4 activity to reduce ROS, decreasing TGF-β1 and inflammatory cytokines containing TNF-α, IL-1β, IL-6 in the lung. | [46] | |
| H2S | NaHS | Renal fibrosis | NaHS suppresses the levels of TGF-β1/Smads and NF-κB signaling pathway. | [83] |
| Renal fibrosis | NaHS inhibits the differentiation of renal fibroblasts into myoblasts by inhibiting TGF-β1/Smads and MAPK signaling pathways. | [82] | ||
| Renal fibrosis | NaHS ameliorates renal fibrosis by inhibition of M1 and M2 macrophage infiltration and NLRP3 activation and subsequently NF-κB and IL-4/STAT6 signaling. | [84] | ||
| Renal fibrosis | NaHS up-regulates the expression of CX40, CX43 and CX45 and regulating MMPs/TIMPs parameters. | [86] | ||
| Renal fibrosis | NaHS improves renal tissue fibrosis by inhibiting autophagy, upregulating SOD and downregulating AKT, TGF-β1 and NF-κB in diabetic rats. | [87] | ||
| Renal fibrosis | NaHS inhibits fibrosis by inhibiting the ERK1/2 pathway and TGF-β1 signaling. | [88] | ||
| Myocardial fibrosis | NaHS modulates MMPs/TIMPs and TGF-β1 expression to attenuate cardiac fibrosis. | [94] | ||
| Myocardial fibrosis | NaHS ameliorates myocardial fibrosis by decreasing myocardial autophagy and activating the PI3K/AKT1 pathway. | [96] | ||
| Myocardial fibrosis | NaHS attenuates myocardial fibrosis in diabetic rats by down-regulating JAK-STAT signaling pathway, thereby inhibiting oxidative stress and ER stress, inflammatory response and apoptosis. | [98] | ||
| Myocardial fibrosis | NaHS ameliorates thyroxin-induced myocardial fibrosis by up-regulating the expression of PI3K/AKT signaling pathway and down-regulating the expression of miR-21, miR-34a and miR-214. | [99] | ||
| Myocardial fibrosis | NaHS inhibits MMPs/TIMPs expression, inflammatory response and alleviates myocardial fibrosis by regulating PKC-ERK1/2MAPK signaling pathway. | [100] | ||
| Myocardial fibrosis | NaHS inhibits myocardial fibrosis by down-regulating the typical Wnt and TGF-β1/Smad3 pathways, reducing myocardial collagen deposition. | [101] | ||
| Myocardial fibrosis | NaHS inhibits doxorubicin-induced myocardial fibrosis by inhibition of overactivation of the ER and that of autophagy via upregulation of the PI3K/AKT/mTOR pathway. | [102] | ||
| Myocardial fibrosis | NaHS delays myocardial fibrosis by increasing the expression of miR-133a, and CTGF. | [103] | ||
| Myocardial fibrosis | NaHS inhibits necroptosis and alleviates hypoxia-induced cardiac fibroblast proliferation via SIRT3. | [104] | ||
| Liver fibrosis | NaHS inhibits the proliferation of activated hsc, induce cell cycle arrest and apoptosis, and weaken CCL4-induced liver fibrosis and ECM expression. | [9] | ||
| Liver fibrosis | NaHS inhibits the formation of liver fibrosis by down-regulating AGTR1 expression. | [107] | ||
| Idiopathic pulmonary fibrosis | NaHS attenuates pulmonary fibrosis possibly through reducing oxidative stress. | [110] | ||
| Idiopathic pulmonary fibrosis | NaHS induces the nuclear accumulation of Nrf2 in lung tissue of smoking rats, thereby up-regulating the expression of antioxidant genes HO-1 and Trx-1 and inhibiting pulmonary fibrosis. | [113] | ||
| H2S gas | Idiopathic pulmonary fibrosis | H2S inhibits the progression of EMT in alveolar epithelial cells and alleviates pulmonary fibrosis by reducing the phosphorylation of Smad2/3. | [111] | |
| Idiopathic pulmonary fibrosis | H2S inhibits fibrosis by inhibiting NF-κB p65 expression and regulating Th1/Th2 balance. | [112] | ||
| NaHS, GYY4137 | Liver fibrosis | The exogenous and endogenous H2S increases the proliferation and activation of HSCs, and suppressed H2S can decrease the proliferation and fibrotic marks of HSCs. | [108] | |
| Hydrogen gas | Idiopathic pulmonary fibrosis | Hydrogen gas inhibits pulmonary fibrosis by reducing the production of ROS and TGF-β. | [119] | |
| Hydrogen | Pulmonary fibrosis | Hydrogen gas restrains the expression of IL-6, IL-4 and IL-13, and suppressing M2-biased macrophage polarization to reduce alveolar fibrosis. | [120] | |
| Myocardial fibrosis | Hydrogen gas ameliorates myocardial fibrosis mainly by inhibiting NLRP3-mediated pyroptosis. | [125] | ||
| Peritoneal fibrosis | Hydrogen gas exerts anti-peritoneal fibrosis effect through inhibiting ROS/PTEN/AKT/mTOR pathway. | [133] | ||
| Myocardial fibrosis | Hydrogen gas decreases the level of ROS and the phosphorylation of p38 MAPK, Smad2/3, the expression of TGF-β1 and CTGF to reduce myocardial fibrosis. | [123] | ||
| Hydrogen Water | Myocardial fibrosis | Hydrogen Water prevents LPS-induced EMT and pulmonary fibrosis by reducing α-SMA production and inhibiting oxidative stress. | [118] | |
| Liver fibrosis | Molecular hydrogen inhibits liver fibrosis by interacting with HO1/IL-10, inhibiting the expression of phosphorylated STAT3 and activation of downstream pMAPK signals. | [128] | ||
| Renal fibrosis | Hydrogen Water reduces the fibrosis and infiltration of macrophage in UUO kidneys by decreasing MDA level and increasing SOD activity. | [130] | ||
| Renal fibrosis | Hydrogen Water reduces renal fibrosis by increasing the expression of Sirt1, a downstream molecule of TGF-β1. | [10] | ||
| Peritoneal fibrosis | Hydrogen Water through inhibiting the expression of TGF-β1 and PAI-1, increasing the expression of t-PA, and reducing oxidative stress and inflammation. | [131] | ||
| SO2 | Na2SO3/NaHSO3 | Myocardial fibrosis | SO2 attenuates myocardial fibrosis in diabetic rats by down-regulating Hippo-MST pathway to reduce apoptosis and ER stress. | [135] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, A.; Wu, S.; Li, Q.; Wang, Q.; Chen, Y. Elucidating the Molecular Pathways and Therapeutic Interventions of Gaseous Mediators in the Context of Fibrosis. Antioxidants 2024, 13, 515. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox13050515
Li A, Wu S, Li Q, Wang Q, Chen Y. Elucidating the Molecular Pathways and Therapeutic Interventions of Gaseous Mediators in the Context of Fibrosis. Antioxidants. 2024; 13(5):515. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox13050515
Chicago/Turabian StyleLi, Aohan, Siyuan Wu, Qian Li, Qianqian Wang, and Yingqing Chen. 2024. "Elucidating the Molecular Pathways and Therapeutic Interventions of Gaseous Mediators in the Context of Fibrosis" Antioxidants 13, no. 5: 515. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox13050515