
Plasma Exchange in Anti-Signal Recognition Particle Myopathy: A Systematic Review and Combined Analysis of Patient Individual Data
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Inclusion Criteria
2.2. Data Sources
2.3. Selection and Data Collection
2.4. Statistical Analysis
3. Results
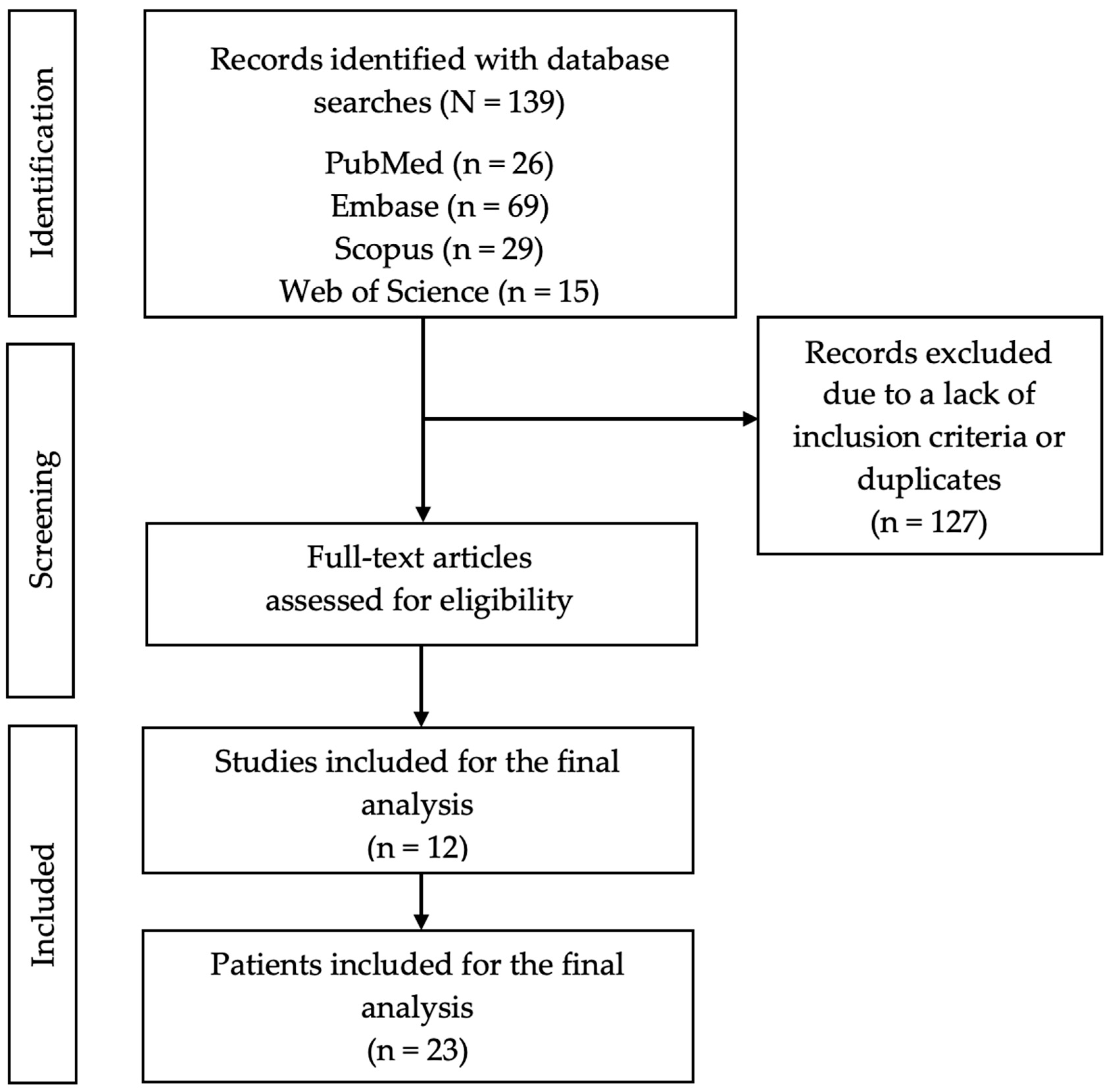
3.1. Study Selection
3.2. Study Characteristics
3.3. Epidemiological Findings
3.4. Clinical Features
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allenbach, Y.; Benveniste, O.; Stenzel, W.; Boyer, O. Immune-mediated necrotizing myopathy: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2020, 16, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Merlonghi, G.; Antonini, G.; Garibaldi, M. Immune-mediated necrotizing myopathy (IMNM): A myopathological challenge. Autoimmun. Rev. 2022, 21, 102993. [Google Scholar] [CrossRef]
- Grau, J.M.; Casademont, J.; Cardellach, F. Enfermedades musculares. In Farreras Rozman Medicina Interna, 18th ed.; Elservier: Amsterdam, The Netherlands, 2016; pp. 1438–1495. [Google Scholar]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Immune-Mediated Necrotizing Myopathy. Curr. Rheumatol. Rep. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Needham, M. Necrotizing autoimmune myopathy. Curr. Opin. Rheumatol. 2011, 23, 612–619. [Google Scholar] [CrossRef]
- Basharat, P.; Christopher-Stine, L. Immune-Mediated Necrotizing Myopathy: Update on Diagnosis and Management. Curr. Rheumatol. Rep. 2015, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Allenbach, Y.; Benveniste, O. Peculiar clinicopathological features of immune-mediated necrotizing myopathies. Curr. Rheumatol. Rep. 2018, 30, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Kasper, D.; Fauci, A.S.; Hauser, A.; Longo, D. Harrison: Principios de Medicina Interna, 19th ed.; McGraw-Hill: New York, NY, USA, 2022. [Google Scholar]
- Lundberg, I.E.; Tjärnlund, A.; Bottai, M.; Werth, V.P.; Pilkington, C.; Visser, M.; Alfredsson, L.; Amato, A.A.; Barohn, R.J.; Liang, M.H.; et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adults and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann. Rheum. Dis. 2017, 76, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Allenbach, Y.; Mammen, A.L.; Benveniste, O.; Stenzel, W.; Immune-Mediated Necrotizing Myopathies Working Group. 224th ENMC International Workshop: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies. Neuromuscul. Disord. 2018, 28, 87–99. [Google Scholar] [CrossRef]
- Goyal, N.A.; Mozaffar, T. Novel Therapeutic Options in Treatment of Idiopathic Inflammatory Myopathies. Curr. Treat. Options Neurol. 2018, 20, 37. [Google Scholar] [CrossRef]
- Dalakas, M.C. Inflammatory Muscle Diseases. N. Engl. J. Med. 2015, 372, 1734–1747. [Google Scholar] [CrossRef]
- Roger, A. Diagnóstico Electromiográfico de las Enfermedades Neuromusculares. Available online: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0138-65572007000100008&lng=es (accessed on 22 April 2024).
- Weeding, E.; Tiniakou, E. Therapeutic Management of Immune-Mediated Necrotizing Myositis. Curr. Treatm. Opt. Rheumatol. 2021, 7, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Arlet, J.B.; Dimitri, D.; Pagnoux, C.; Boyer, O.; Maisonobe, T.; Authier, F.J.; Bloch-Queyrat, C.; Goulvestre, C.; Heshmati, F.; Atassi, M.; et al. Marked efficacy of a therapeutic strategy associating prednisone and plasma exchange followed by rituximab in two patients with refractory myopathy associated with antibodies to the signal recognition particle (SRP). Neuromuscul. Disord. 2006, 16, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Komaki, H.; Saito, T.; Saito, Y.; Nakagawa, E.; Sugai, K.; Sasaki, M.; Hayashi, Y.K.; Nishino, I.; Momomura, M.; et al. A pediatric patient with myopathy associated with antibodies to a signal recognition particle. Brain Dev. 2012, 34, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Valiyil, R.; Casciola-Rosen, L.; Hong, G.; Mammen, A.; Christopher-Stine, L. Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: A case series. Arthritis Care Res. 2010, 62, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Thiébaut, M.; Terrier, B.; Menacer, S.; Berezne, A.; Bussone, G.; Goulvestre, C.; Bellance, R.; Guillevin, L.; Vignaux, O.; Mouthon, L. Antisignal recognition particle antibodies-related cardiomyopathy. Circulation 2013, 127, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Wantke, F.; Kneussl, M.; Hubner, M.; Derfler, K.; Brücke, T.; Schmaldienst, S. Signal recognition particle (SRP) positive myositis in a patient with cryptogenic organizing pneumonia (COP). Rheumatol. Int. 2010, 30, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Momomura, M.; Miyamae, T.; Nozawa, T.; Kikuchi, M.; Kizawa, T.; Imagawa, T.; Drouot, L.; Jouen, F.; Boyer, O.; Yokota, S. Serum levels of anti-SRP54 antibodies reflect disease activity of necrotizing myopathy in a child treated effectively with combinatorial methylprednisolone pulses and plasma exchanges followed by intravenous cyclophosphamide. Mod. Rheumatol. 2014, 24, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Tiwana, H.; Kaur, D. Anti-SRP myopathy in 2 different ethnic groups. J. Clin. Neuromuscul. Dis. 2018, 19, 164–165. [Google Scholar]
- Kamo, H.; Hatano, T.; Kanai, K.; Aoki, N.; Kamiyama, D.; Yokoyama, K.; Takanashi, M.; Yamashita, Y.; Shimo, Y.; Hattori, N. Pembrolizumab-related systemic myositis involving ocular and hindneck muscles resembling myasthenic gravis: A case report. BMC Neurol. 2019, 19, 184. [Google Scholar] [CrossRef]
- Harada, Y.; Guntrum, D.; Veerapandiyan, A.; Mammen, A.; Herrmann, D. Successful recovery of anti-SRP myopathy with subcutaneous methotrexate after 17 years of poor response to immunomodulation. Neurology 2020, 94, 1139. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, Y.; Liu, H.; Cheng, X.; Ye, J.; Hu, Q.; Jia, J.; Wang, M.; Liu, T.; Zhou, Z.; et al. Plasma exchange therapy in refractory inflammatory myopathy with anti-signal recognition particle antibody: A case series. Rheumatology 2022, 61, 2625–2630. [Google Scholar] [CrossRef] [PubMed]
- Savey, L.; Bussone, G.; Lannuzel, A.; Goulvestre, C.; Guillevin, L.; Mouthon, L. Myopathie nécrosante avec auto-anticorps anti-SRP: Efficacité transitoire d’une stratégie thérapeutique associant échanges plasmatiques et rituximab. Presse Med. 2012, 41, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Almaghrabi, M.; Almatrafi, S.B.; Alzahrani, A.; Alharbi, M. Marked Efficacy of a Therapeutic Strategy in a Patient With Necrotizing Myopathy Associated With Anti-signal Recognition Particle (SRP) Autoantibodies. Cureus 2023, 15, e35471. [Google Scholar] [CrossRef]
- Pinto, A.A.; De Seze, J.; Jacob, A.; Reddel, S.; Yudina, A.; Tan, K. Comparison of IVIg and TPE efficacy in the treatment of neurological disorders: A systematic literature review. Ther. Adv. Neurol. Disord. 2023, 16, 17562864231154306. [Google Scholar] [CrossRef] [PubMed]
- Pavlekovics, M.; Engh, M.A.; Lugosi, K.; Szabo, L.; Hegyi, P.; Terebessy, T.; Csukly, G.; Molnar, Z.; Illes, Z.; Lovas, G. Plasma Exchange versus Intravenous Immunoglobulin in Worsening Myasthenia Gravis: A Systematic Review and Meta-Analysis with Special Attention to Faster Relapse Control. Biomedicines 2023, 11, 3180. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, F.; Niu, Y.; Yan, L.; Li, J.; Tan, B.; Qin, L. Therapeutic plasma exchange-free treatment for first-episode TTP: A systematic review. Transfus. Apher. Sci. 2023, 62, 103661. [Google Scholar] [CrossRef] [PubMed]
- Della-Marina, A.; Pawlitzki, M.; Ruck, T.; van-Baalen, A.; Vogt, N.; Schweiger, B.; Hertel, S.; Kölbel, H.; Wiendl, H.; Preuße, C.; et al. Clinical course, myopathology and challenge of therapeutic intervention in pediatric patients with autoimmune-mediated necrotizing myopathy. Children 2021, 8, 721. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Langguth, D.; Hardy, T.A.; Garg, N.; Bundell, C.; Rojana-Udomsart, A.; Dale, R.C.; Robertson, T.; Mammen, A.L.; Reddel, S.W. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e96. [Google Scholar] [CrossRef] [PubMed]
- Bergua, C.; Chiavelli, H.; Allenbach, Y.; Arouche-Delaperche, L.; Arnoult, C.; Bourdenet, G.; Jean, L.; Zoubairi, R.; Guerout, N.; Mahler, M.; et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann. Rheum. Dis. 2019, 78, 131–139. [Google Scholar] [CrossRef]
- Lv, C.; You, H.; Xu, L.; Wang, L.; Yuan, F.; Li, J.; Wu, M.; Zhou, S.; Da, Z.; Qian, J.; et al. Coexistence of Anti-Ro52 Antibodies in Anti-MDA5 Antibody–Positive Dermatomyositis Is Highly Associated with Rapidly Progressive Interstitial Lung Disease and Mortality Risk. J. Rheumatol. 2023, 50, 219–226. [Google Scholar] [CrossRef]
- Nayebirad, S.; Mohamadi, A.; Yousefi-Koma, H.; Javadi, M.; Farahmand, K.; Atef-Yekta, R.; Tamartash, Z.; Jameie, M.; Mohammadzadegan, A.M.; Kavosi, H. Association of anti-Ro52 autoantibody with interstitial lung disease in autoimmune diseases: A systematic review and meta-analysis. BMJ Open Respir. Res. 2023, 10, e002076. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Current review | |
|---|---|
| PE | |
| n = 23 (%) | |
| Sex female/male | 7 (30.4)/16 (69.6) |
| Age at diagnosis, mean years (SD) | |
| Males | 50.1 (22.1) |
| Females | 38.9 (19.9) |
| Total | 42.4 (20.8) |
| Race | |
| Asiatic | 2 (8.7) |
| Caucasian | 3 (13) |
| Afro-American | 2 (8.7) |
| African | 2 (8.7) |
| Not available | 14 (60.9) |
| Initial symptoms | |
| Muscle weakness | 23 (100) |
| Myalgia | 6 (26.1) |
| Dysphagia | 7 (30.4) |
| Anorexia | 1 (4.3) |
| Dyspnea | 3 (13) |
| Asthenia | 2 (8.7) |
| Two or more | 18 (78.3) |
| extramuscular manifestations at disease onset | |
| Yes/No | 11 (47.8)/12 (52.2) |
| Pulmonary | 7 (30.4) |
| Dermatological | 7 (30.4) |
| Raynaud phenomenon | 1 (4.3) |
| Cardiological | 2 (8.7) |
| Joints | 0 |
| Two or more | 5 (45.5) |
| Co-diagnosis with other disease | |
| Yes/No/Unknown | 1 (4.3)/22 (95.7) |
| Neoplasic | 1 (100) |
| Autoimmune | 0 |
| Anti-SRP antibodies documented | |
| At disease onset | |
| Positive | 22 (95.7) |
| Negative | 1 (4.3) |
| Not available | 0 |
| After treatment | |
| Positive | 3 (13) |
| Negative | 1 (4.3) |
| Not available | 19 (82.6) |
| Anti-Ro52 antibodies at disease onset | |
| Positive | 8 (34.8) |
| Negative | 9 (39.1) |
| Not available | 6 (26.1) |
| Other auto-antibodies | |
| Anti-MDA5 | 2 (8.7) |
| Anti-NXP2 | 1 (4.3) |
| Anti-PM-Scl 75 | 1 (4.3) |
| Anti-Mi2 alpha | 1 (4.3) |
| Anti-Yo | 1 (4.3) |
| Anti-dsDNA | 1 (4.3) |
| Creatine kinase (IU/L) | |
| Before treatment; SD | 8586.6; 886.2 |
| After treatment; SD | 573.3; 533.5 |
| Aldolase (IU/L) | |
| Before treatment; SD | 141.9; 84.5 |
| After treatment; SD | Not available |
| Lactate dehydrogenase (IU/L) | |
| Before treatment; SD | 847.4; 311 |
| After treatment; SD | Not available |
| Muscle biopsy | |
| Yes/No | 12 (52.2)/11 (47.8) |
| Compatible/Not compatible | 12 (100)/0 |
| Neuromuscular study | |
| Yes/No/Unknown | 19 (82.6)/2 (8.7)/2 (8.7) |
| Compatible/Not compatible | 19 (100)/0 |
| Treatment before PE | |
| Glucocorticoids (any) | 22 (95.7) |
| Intravenous pulses | 4 (17.4) |
| Methotrexate | 3 (13) |
| Azathioprine | 5 (21.7) |
| Tacrolimus | 4 (17.4) |
| Cyclosporine | 1 (4.3) |
| Mofetil mycophenolate | 2 (8.7) |
| Cyclophosphamide | 4 (17.4) |
| Rituximab | 3 (13) |
| Intravenous immunoglobulins | 13 (56.5) |
| Two combined drugs | 12 (52.2) |
| Three or more combined drugs | 9 (39.1) |
| Response to PE | |
| Complete disease remission | 10 (43.5) |
| Clinical improvement (any) | 10 (43.5) |
| Null response | 2 (8.7) |
| Not available | 1 (4.3) |
| Adverse effects after PE | |
| Yes/No | 2 (8.7)/21 (91.3) |
| Allergic | 1 (4.3) |
| Vascular | 1 (4.3) |
| Relapses after PE | |
| Yes/No/Unknown | |
| Adults | 4 (20)/13 (65)/3 (15) |
| Under 18 years old | 0/3 (100)/0 |
| Disease-free interval in months, mean; SD | 76; 83.5 |
| Number of relapses, mean; SD | 3.25; 1.9 |
| Time until remission, months; SD | 31.6; 63 |
| Deaths | 3 (13) |
| Before PE | After PE | |||
|---|---|---|---|---|
| n/N (%) | n/N (%) | p Value | Response Rate (%) | |
| Clinical manifestations | ||||
| Muscle weakness | 20/20 (100) | 9/20 (45) | 0.001 | 11/20 (55) |
| Myalgia | 3/12 (25) | 0/12 (0) | 0.250 | 3/3 (100) |
| Dysphagia | 5/12 (41.7) | 0/12 (0) | 0.063 | 5/5 (100) |
| Anorexia | 1/12 (8.3) | 0/12 (0) | 1 | 1/1 (100) |
| Dyspnea | 3/12 (25) | 0/12 (0) | 0.250 | 3/3 (100) |
| Asthenia | 2/12 (16.7) | 0/12 (0) | 0.500 | 2/2 (100) |
| Before PE | After PE | ||
|---|---|---|---|
| n/N (%) | n/N (%) | p Value | |
| Corticosteroids | 10/11 (90.9) | 10/11 (90.9) | 1 |
| Pulsed corticosteroids | 3/11 (27.3) | 0/11 | 0.250 |
| Additional immunosuppressant | 11/13 (84.6) | 10/13 (76.9) | 1 |
| Corticosteroids sparing agents | 5/12 (41.7) | 7/12 (58.3) | 0.687 |
| Methotrexate | 2/11 (18.2) | 4/11 (36.4) | 0.500 |
| Tacrolimus | 0/12 (0) | 0/12 (0) | 1 |
| Ciclosporin | 1/12 (8.3) | 1/12 (8.3) | 1 |
| Mofetil mycophenolate | 2/12 (16.7) | 1/12 (8.3) | 1 |
| Cyclophosphamide | 3/12 (25.0) | 2/12 (16.7) | 1 |
| Azathioprine | 4/11 (36.4) | 3/11 (27.3) | 1 |
| Rituximab | 3/13 (23.1) | 3/13 (23.1) | 1 |
| Intravenous immunoglobulins | 7/12 (58.3) | 0/12 (0) | 0.016 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Rodríguez, P.; Escribano-Iglesias, M.; Crisolino-Pozas, Á.-P.; Cubino-Boveda, N.; López-Parra, M.; Marcos, M.; Chamorro, A.-J. Plasma Exchange in Anti-Signal Recognition Particle Myopathy: A Systematic Review and Combined Analysis of Patient Individual Data. J. Pers. Med. 2024, 14, 461. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm14050461
Martínez-Rodríguez P, Escribano-Iglesias M, Crisolino-Pozas Á-P, Cubino-Boveda N, López-Parra M, Marcos M, Chamorro A-J. Plasma Exchange in Anti-Signal Recognition Particle Myopathy: A Systematic Review and Combined Analysis of Patient Individual Data. Journal of Personalized Medicine. 2024; 14(5):461. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm14050461
Chicago/Turabian StyleMartínez-Rodríguez, Pablo, María Escribano-Iglesias, Ángel-P. Crisolino-Pozas, Noelia Cubino-Boveda, Miriam López-Parra, Miguel Marcos, and Antonio-J. Chamorro. 2024. "Plasma Exchange in Anti-Signal Recognition Particle Myopathy: A Systematic Review and Combined Analysis of Patient Individual Data" Journal of Personalized Medicine 14, no. 5: 461. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm14050461