

Bacteroidales-Specific Antimicrobial Genes Can Influence the Selection of the Dominant Fecal Strain of Bacteroides vulgatus and Bacteroides uniformis from the Gastrointestinal Tract Microbial Community

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Publicly Available Data Sets Were Used in This Study
2.2. Analysis of Bacteroides BSAP Genes
2.3. Analysis of Bacteroides spp. Strain Relatedness Using WSS
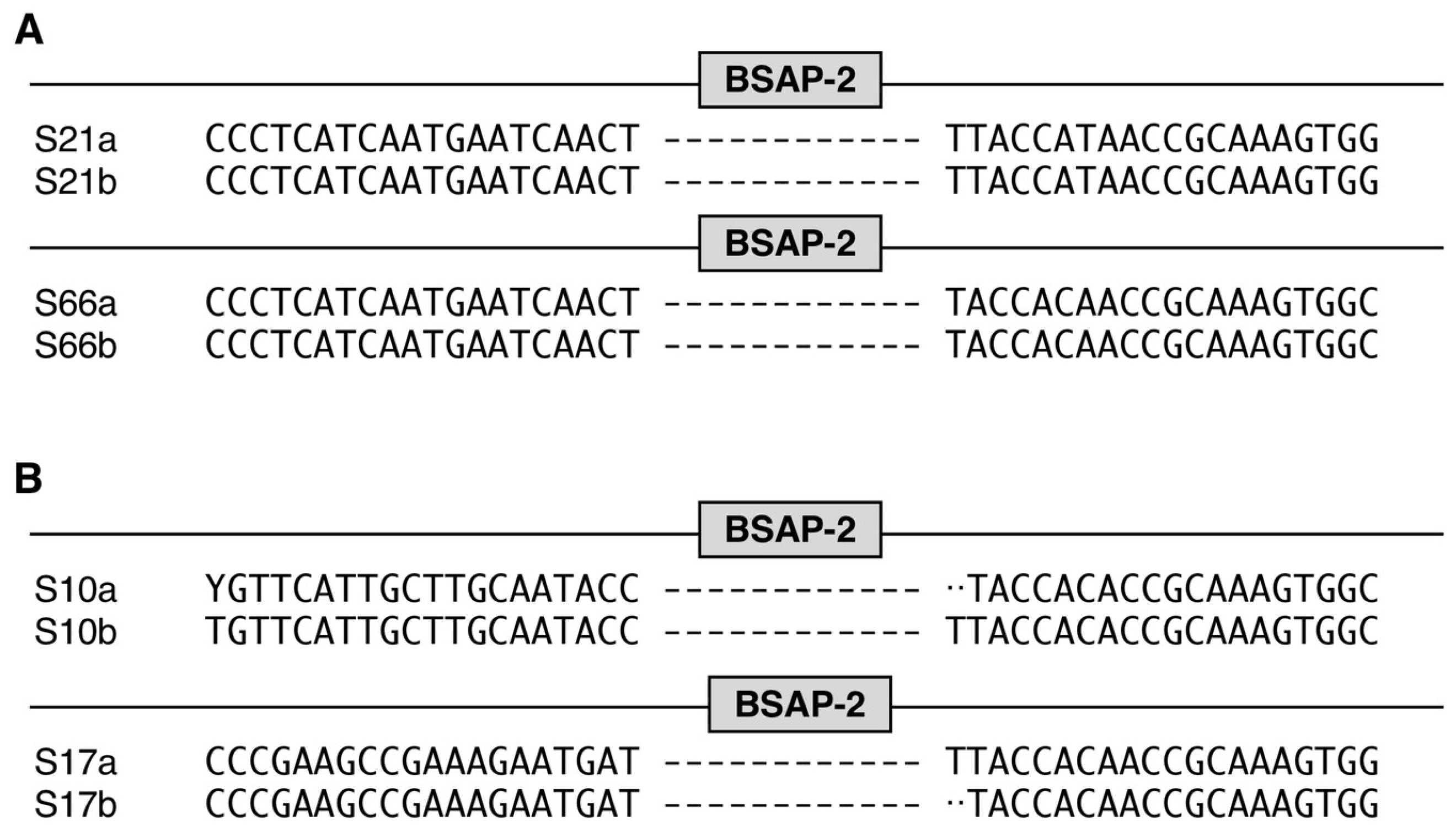
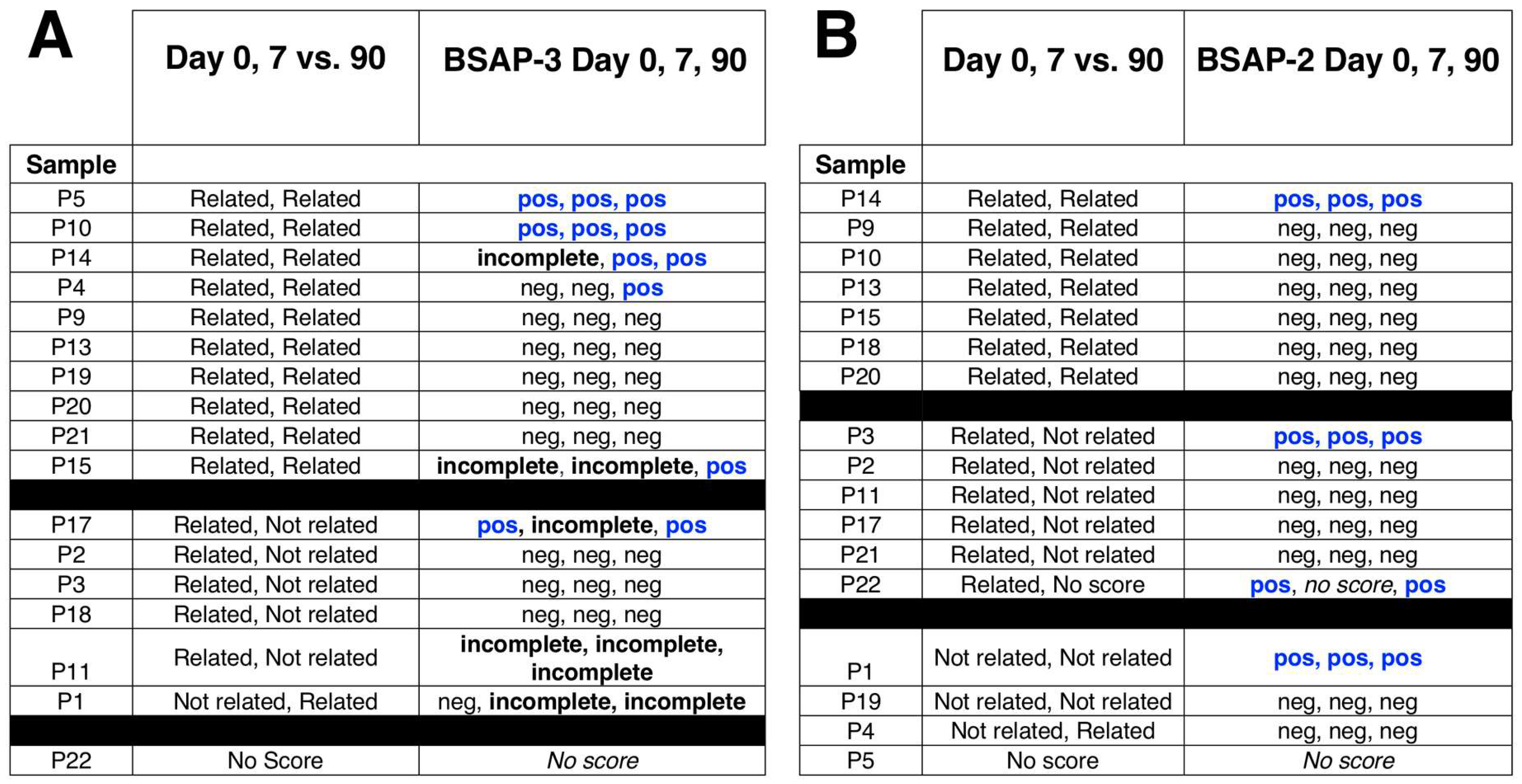
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schloissnig, S.; Arumugam, M.; Sunagawa, S.; Mitreva, M.; Tap, J.; Zhu, A.; Waller, A.; Mende, D.R.; Kultima, J.R.; Martin, J.; et al. Genomic variation landscape of the human gut microbiome. Nature 2013, 493, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Yi, N.; Zhi, D.; Eipers, P.; Goldsmith, K.T.; Dixon, P.; Crossman, D.K.; Crowley, M.R.; Lefkowitz, E.J.; Rodriguez, J.M.; et al. Identification of donor microbe species that colonize and persist long term in the recipient after fecal transplant for recurrent Clostridium difficile. NPJ Biofilms Microbiomes 2017, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef]
- Koo, H.; Hakim, J.A.; Crossman, D.K.; Lefkowitz, E.J.; Morrow, C.D. Sharing of gut microbial strains between selected individual sets of twins cohabitating for decades. PLoS ONE 2019, 14, e0226111. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.-U.; Zaura, E.; Buijs, M.J.; Keijser, B.J.F.; Crielaard, W.; Nord, C.E.; Weintraub, A. Determining the long-term effect of antibiotic administration on the human normal intestinal microbiota using culture and pyrosequencing methods. Clin. Infect. Dis. 2015, 60, S77–S84. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Hakim, J.A.; Crossman, D.K.; Kumar, R.; Lefkowitz, E.J.; Morrow, C.D. Individualized recovery of gut microbial strains post antibiotics. NPJ Biofilms Microbiomes 2019, 5, 30. [Google Scholar] [CrossRef]
- Roodgar, M.; Good, B.H.; Garud, N.R.; Martis, S.; Avula, M.; Zhou, W.; Lancaster, S.M.; Lee, H.; Babveyh, A.; Nesamoney, S.; et al. Longitudinal linked-read sequencing reveals ecological and evolutionary responses of a human gut microbiome during antibiotic treatment. Genome Res. 2021, 31, 1433–1446. [Google Scholar] [CrossRef]
- Wexler, A.G.; Goodman, A.L. An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol. 2017, 2, 17026. [Google Scholar] [CrossRef]
- Kraal, L.; Abubucker, S.; Kota, K.; Fischbach, M.A.; Mitreva, M. The prevalence of species and strains in the human microbiome: A resource for experimental efforts. PLoS ONE 2014, 9, e97279. [Google Scholar] [CrossRef]
- Koo, H.; Crossman, D.K.; Morrow, C.D. Strain Tracking to Identify Individualized Patterns of Microbial Strain Stability in the Developing Infant Gut Ecosystem. Front. Pediatr. 2020, 8, 549844. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; McFarland, B.C.; Hakim, J.A.; Crossman, D.K.; Crowley, M.R.; Rodriguez, J.M.; Benveniste, E.N.; Morrow, C.D. An individualized mosaic of maternal microbial strains is transmitted to the infant gut microbial community. R. Soc. Open Sci. 2020, 7, 192200. [Google Scholar] [CrossRef] [PubMed]
- McEneany, V.L.; Coyne, M.J.; Chatzidaki-Livanis, M.; Comstock, L.E. Acquisition of MACPF domain-encoding genes is the main contributor to LPS glycan diversity in gut Bacteroides species. ISME J. 2018, 12, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Matano, L.M.; Coyne, M.J.; García-Bayona, L.; Comstock, L.E. Bacteroidetocins Target the Essential Outer Membrane Protein BamA of Bacteroidales Symbionts and Pathogens. mBio 2021, 12, e02221–e02285. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Pop, M.; Creasy, H.H.; Giglio, M.G.; Huttenhower, C.; Gevers, D.; Petrosino, J.F.; Abubucker, S.; Badger, J.H.; et al. A framework for human microbiome research. Nature 2012, 486, 215. [Google Scholar]
- Raymond, F.; Ouameur, A.A.; Déraspe, M.; Iqbal, N.; Gingras, H.; Dridi, B.; Leprohon, P.; Plante, P.-L.; Giroux, R.; Bérubé, È.; et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 2016, 10, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Grams, J.; Chu, D.I.; Crossman, D.K.; Stahl, R.; Eipers, P.; Goldsmith, K.; Crowley, M.; Lefkowitz, E.J.; Morrow, C.D. New microbe genomic variants in patients fecal community following surgical disruption of the upper human gastrointestinal tract. Hum. Microbiome J. 2018, 10, 37–42. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, K.G.; Coyne, M.J.; Gentyala, R.R.; Chatzidaki-Livanis, M.; Comstock, L.E. Bacteroidales secreted antimicrobial proteins target surface molecules necessary for gut colonization and mediate competition in vivo. mBio 2016, 7, e01055-16. [Google Scholar] [CrossRef]
- Salyers, A.A.; Shoemaker, N.B.; Stevens, A.M.; Li, L.-Y. Conjugative transposons: An unusual and diverse set of integrated gene transfer elements. Microbiol. Rev. 1995, 59, 579–590. [Google Scholar] [CrossRef]
- Cheng, Q.; Paszkiet, B.J.; Shoemaker, N.B.; Gardner, J.F.; Salyers, A.A. Integration and excision of a Bacteroides conjugative transposon, CTnDOT. J. Bacteriol. 2000, 182, 4035–4043. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.A.; Waldor, M.K. Integrative and conjugative elements: Mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010, 8, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Coyne, M.J.; Zitomersky, N.L.; McGuire, A.M.; Earl, A.M.; Comstock, L.E. Evidence of extensive DNA transfer between bacteroidales species within the human gut. mBio 2014, 5, e01305–e01314. [Google Scholar] [CrossRef] [PubMed]
- Capy, P.; Gasperi, G.; Biémont, C.; Bazin, C. Stress and transposable elements: Co-evolution or useful parasites? Heredity 2000, 85, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Twiss, E.; Coros, A.M.; Tavakoli, N.P.; Derbyshire, K.M. Transposition is modulated by a diverse set of host factors in Escherichia coli and is stimulated by nutritional stress. Mol. Microbiol. 2005, 57, 1593–1607. [Google Scholar] [CrossRef]
- Maharjan, R.P.; Ferenci, T. A shifting mutational landscape in 6 nutritional states: Stress-induced mutagenesis as a series of distinct stress input–mutation output relationships. PLoS Biol. 2017, 15, e2001477. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Morrow, C.D. Perturbation of the human gastrointestinal tract microbial ecosystem by oral drugs to treat chronic disease results in a spectrum of individual specific patterns of extinction and persistence of dominant microbial strains. PLoS ONE 2020, 15, e0242021. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Morrow, C.D. Time series strain tracking analysis post fecal transplantation identifies individual specific patterns of fecal dominant donor, recipient, and unrelated microbial strains. PLoS ONE 2022, 17, e0274633. [Google Scholar] [CrossRef]
- Koo, H.; Morrow, C.D. Identification of donor Bacteroides vulgatus genes encoding proteins that correlate with early colonization following fecal transplant of patients with recurrent Clostridium difficile. Sci. Rep. 2023, 13, 14112. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koo, H.; Morrow, C.D. Bacteroidales-Specific Antimicrobial Genes Can Influence the Selection of the Dominant Fecal Strain of Bacteroides vulgatus and Bacteroides uniformis from the Gastrointestinal Tract Microbial Community. Life 2024, 14, 555. https://0-doi-org.brum.beds.ac.uk/10.3390/life14050555
Koo H, Morrow CD. Bacteroidales-Specific Antimicrobial Genes Can Influence the Selection of the Dominant Fecal Strain of Bacteroides vulgatus and Bacteroides uniformis from the Gastrointestinal Tract Microbial Community. Life. 2024; 14(5):555. https://0-doi-org.brum.beds.ac.uk/10.3390/life14050555
Chicago/Turabian StyleKoo, Hyunmin, and Casey D. Morrow. 2024. "Bacteroidales-Specific Antimicrobial Genes Can Influence the Selection of the Dominant Fecal Strain of Bacteroides vulgatus and Bacteroides uniformis from the Gastrointestinal Tract Microbial Community" Life 14, no. 5: 555. https://0-doi-org.brum.beds.ac.uk/10.3390/life14050555