
Liver X Receptor Ligand GAC0001E5 Downregulates Antioxidant Capacity and ERBB2/HER2 Expression in HER2-Positive Breast Cancer Cells

Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Treatments, Cell Lines, and Culture
2.2. Cell Proliferation Assay
2.3. Real-Time Quantitative PCR
2.4. Western Blot Analysis
2.5. IC50 Assays
2.6. Lapatinib+1E5 Combination Treatment
2.7. FASN Inhibitor Analysis
2.8. Glutamine Dependency Assay
2.9. Intracellular GSH/GSSG Ratio
2.10. ROS Levels
2.11. TCGA Clinical Data Analysis
3. Results
3.1. Liver X Receptor Ligand 1E5 Disrupts HER2-Positive Breast Cancer Cell Viability
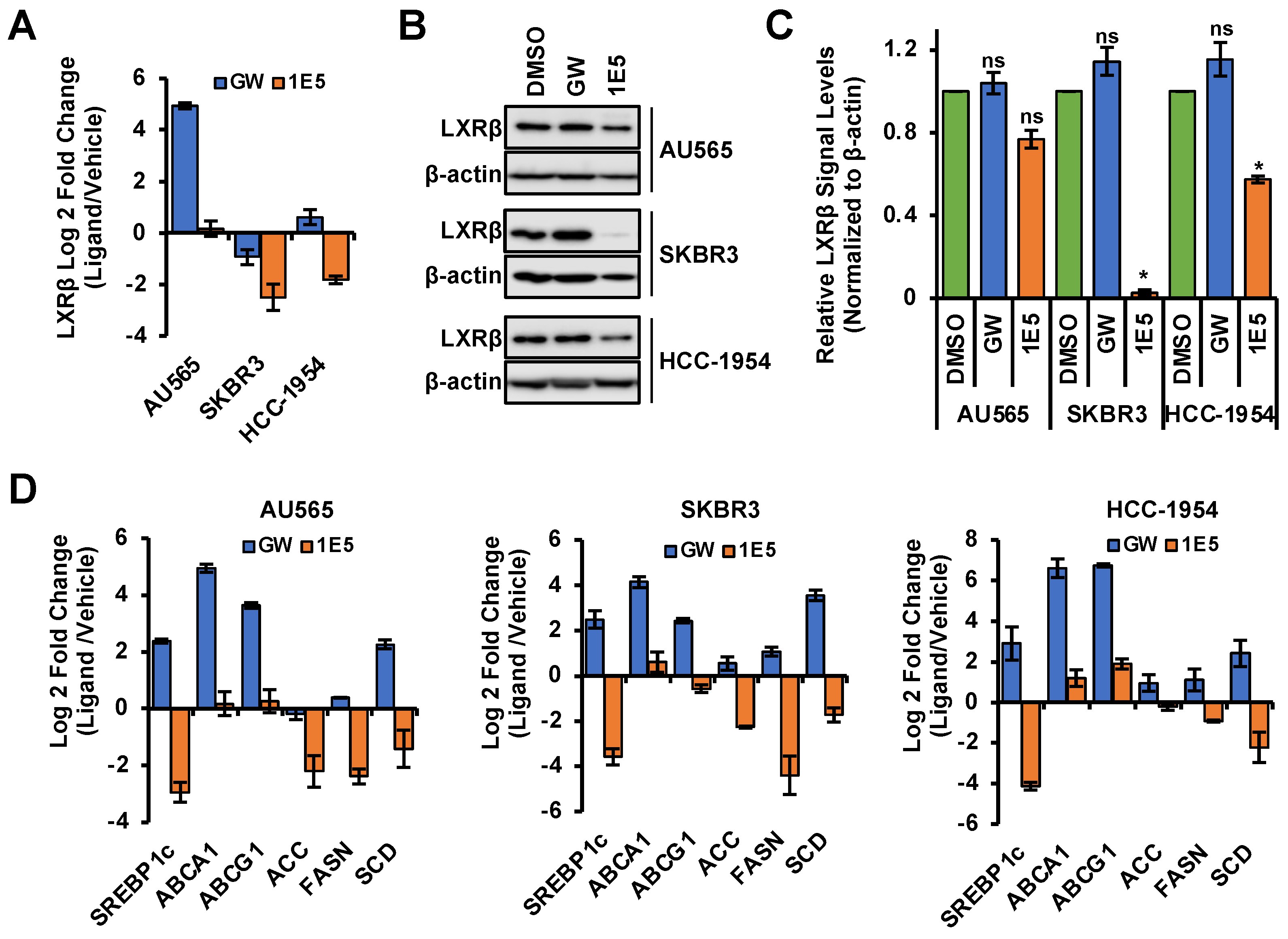
3.2. 1E5 Is an LXR Inverse Agonist in HER2-Positive Breast Cancer Cells
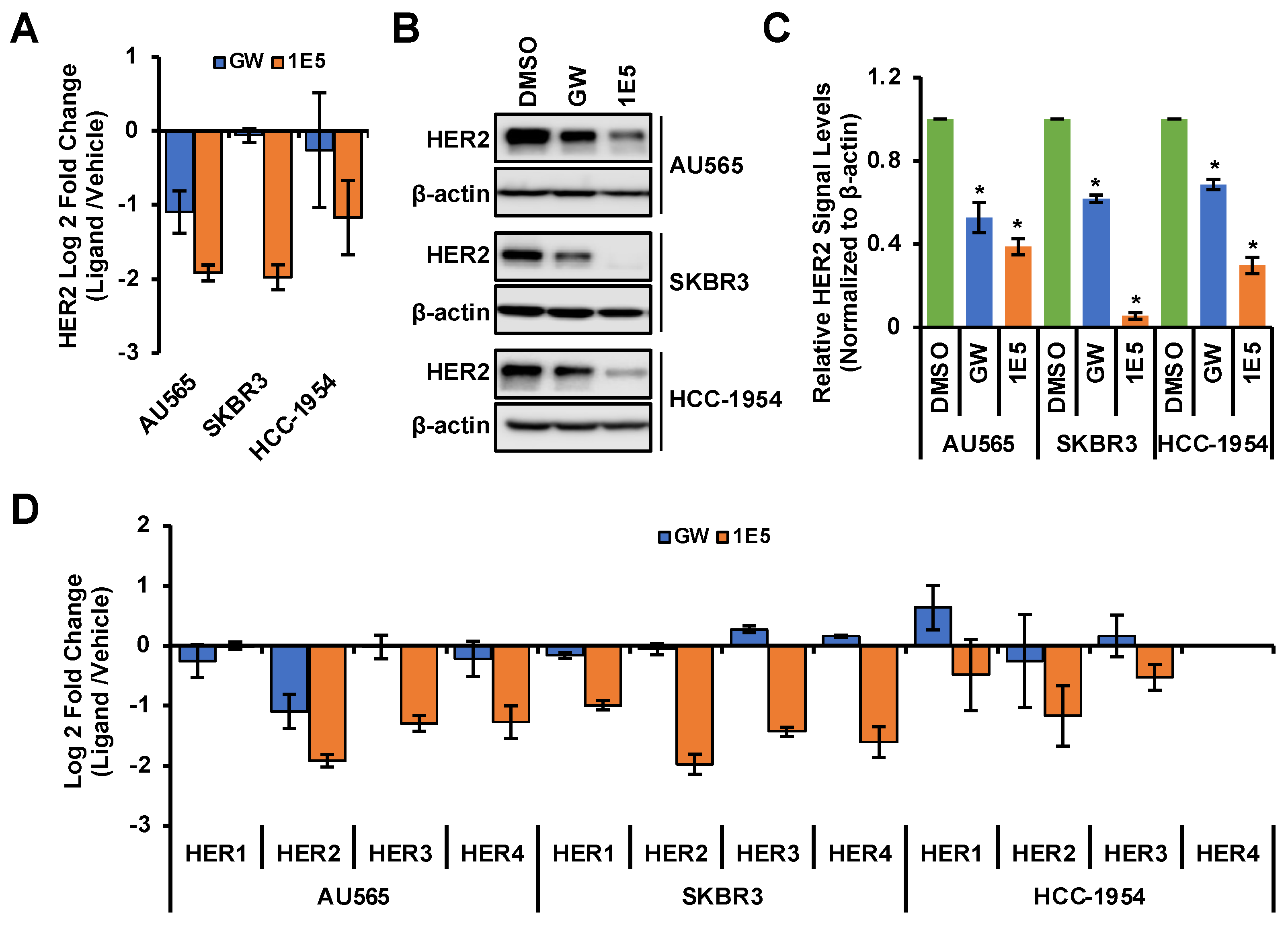
3.3. HER2 Expression Is Downregulated by 1E5
3.4. Additive Effects of 1E5 and Lapatinib Suggest Complementary Targeting of HER2
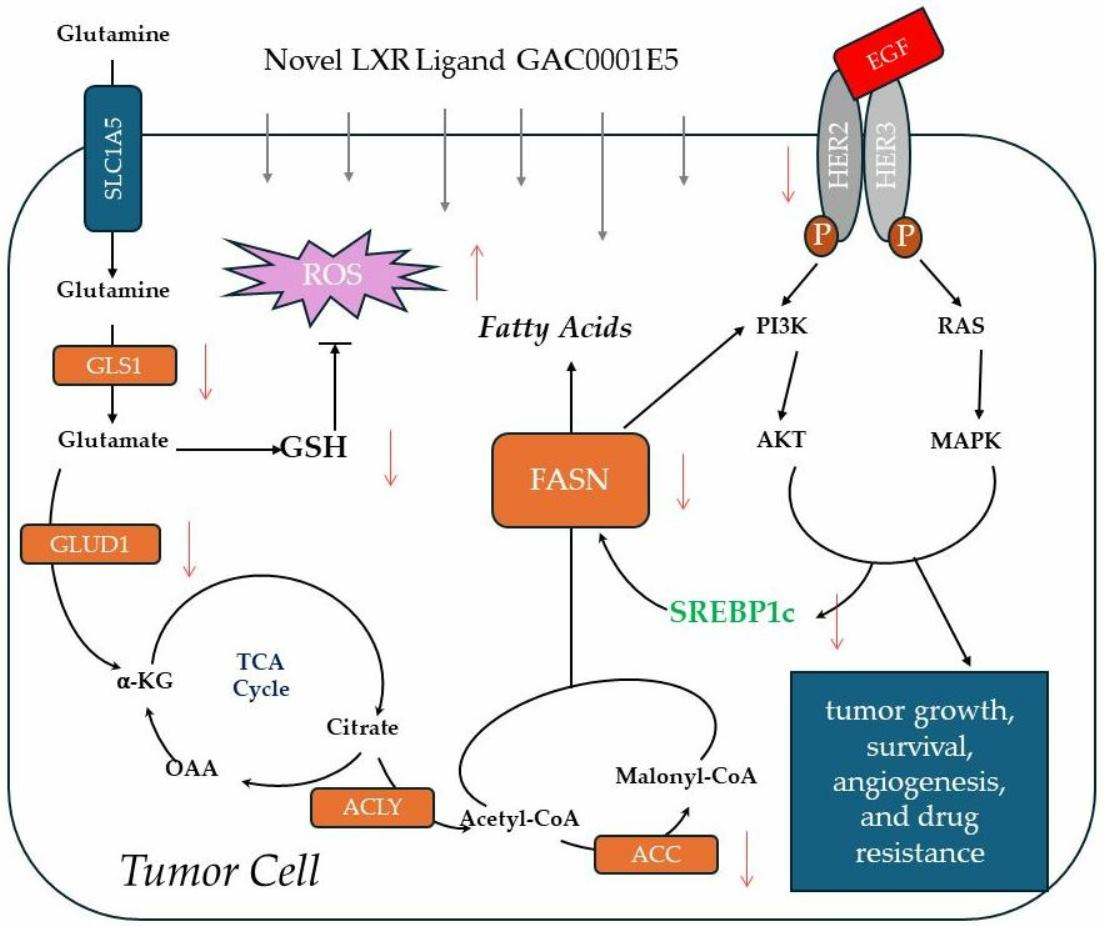
3.5. Expression of Fatty Acid Synthesis Genes Is Disrupted by 1E5
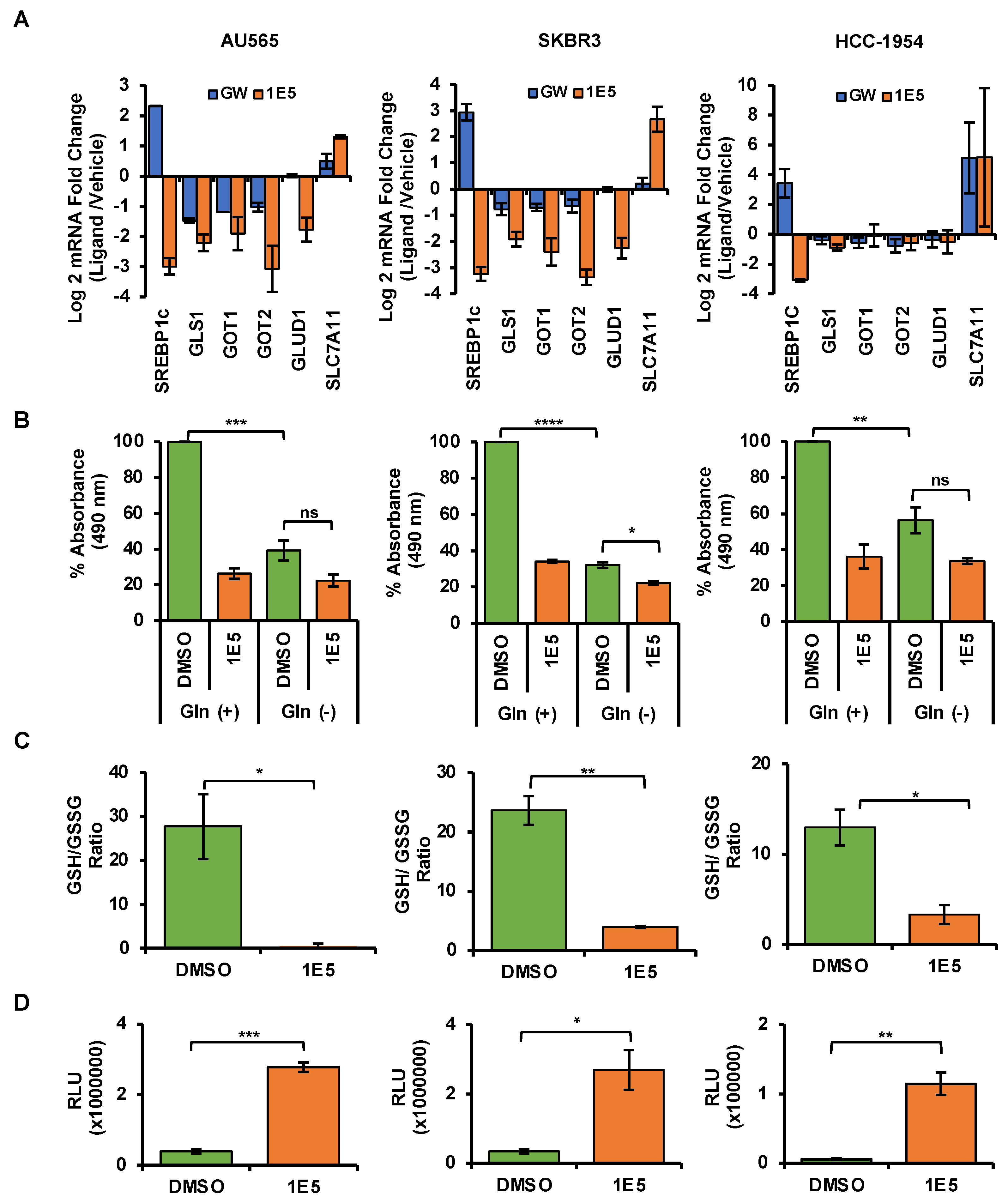
3.6. LXR Ligand 1E5 Suppresses Glutaminolysis and Induces Oxidative Stress
3.7. Lipogenesis Genes Downregulated by 1E5 Are Overexpressed in HER2-Positive Breast Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Masood, S. Breast Cancer Subtypes: Morphologic and Biologic Characterization. Women’s Health 2016, 12, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef]
- Scerri, J.; Scerri, C.; Schäfer-Ruoff, F.; Fink, S.; Templin, M.; Grech, G. PKC-mediated phosphorylation and activation of the MEK/ERK pathway as a mechanism of acquired trastuzumab resistance in HER2-positive breast cancer. Front. Endocrinol. 2022, 13, 1010092. [Google Scholar] [CrossRef]
- Patel, A.; Unni, N.; Peng, Y. The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer. Cancers 2020, 12, 2081. [Google Scholar] [CrossRef] [PubMed]
- Tapia, M.; Hernando, C.; Martínez, M.T.; Burgués, O.; Tebar-Sánchez, C.; Lameirinhas, A.; Ágreda-Roca, A.; Torres-Ruiz, S.; Garrido-Cano, I.; Lluch, A.; et al. Clinical Impact of New Treatment Strategies for HER2-Positive Metastatic Breast Cancer Patients with Resistance to Classical Anti-HER Therapies. Cancers 2023, 15, 4522. [Google Scholar] [CrossRef] [PubMed]
- Prete, S.D.; Montella, L.; Arpino, G.; Buono, G.; Buonerba, C.; Dolce, P.; Fiorentino, O.; Aliberti, M.; Febbraro, A.; Savastano, C.; et al. Second line trastuzumab emtansine following horizontal dual blockade in a real-life setting. Oncotarget 2020, 11, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Yu, Q.; Liu, Z.; Li, C.; Wang, F.; Yu, Z. The Effectiveness of Lapatinib in HER2-Positive Metastatic Breast Cancer Patients Pretreated With Multiline Anti-HER2 Treatment: A Retrospective Study in China. Technol. Cancer Res. Treat. 2021, 20, 153303382110378. [Google Scholar] [CrossRef]
- Schlam, I.; Tarantino, P.; Tolaney, S.M. Overcoming Resistance to HER2-Directed Therapies in Breast Cancer. Cancers 2022, 14, 3996. [Google Scholar] [CrossRef]
- Sever, R.; Glass, C.K. Signaling by Nuclear Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef]
- Scholtes, C.; Giguère, V. Transcriptional control of energy metabolism by nuclear receptors. Nat. Rev. Mol. Cell Biol. 2022, 23, 750–770. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Gustafsson, J.-Å. Targeting liver X receptors in cancer therapeutics. Nat. Rev. Cancer 2015, 15, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Vu, T.; Vedin, L.-L.; Liu, K.; Jonsson, P.; Lin, J.Z.; Candelaria, N.R.; Candelaria, L.P.; Addanki, S.; Williams, C.; Gustafsson, J.-Å.; et al. Liver × receptor ligands disrupt breast cancer cell proliferation through an E2F-mediated mechanism. Breast Cancer Res. 2013, 15, R51. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, N.R.; Addanki, S.; Zheng, J.; Nguyen-Vu, T.; Karaboga, H.; Dey, P.; Gabbi, C.; Vedin, L.-L.; Liu, K.; Wu, W.; et al. Antiproliferative Effects and Mechanisms of Liver X Receptor Ligands in Pancreatic Ductal Adenocarcinoma Cells. PLoS ONE 2014, 9, e106289. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Vedin, L.-L.; Steffensen, K.R. The emerging roles of liver X receptors and their ligands in cancer. Expert Opin. Ther. Targets 2016, 20, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.P.; Schmidt, R.J.; Foxworthy, P.; Zhang, Y.; Dai, J.; Bensch, W.R.; Kauffman, R.F.; Gao, H.; Ryan, T.P.; Jiang, X.-C.; et al. Coadministration of a Liver X Receptor Agonist and a Peroxisome Proliferator Activator Receptor-α Agonist in Mice: Effects of Nuclear Receptor Interplay on High-Density Lipoprotein and Triglyceride Metabolism in Vivo. J. Pharmacol. Exp. Ther. 2004, 309, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Karaboga, H.; Huang, W.; Srivastava, S.; Widmann, S.; Addanki, S.; Gamage, K.T.; Mazhar, Z.; Ebalunode, J.O.; Briggs, J.M.; Gustafsson, J.-Å.; et al. Screening of Focused Compound Library Targeting Liver X Receptors in Pancreatic Cancer Identified Ligands with Inverse Agonist and Degrader Activity. ACS Chem. Biol. 2020, 15, 2916–2928. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Widmann, S.; Ho, C.; Nguyen, D.; Nguyen, A.; Premaratne, A.; Gustafsson, J.-Å.; Lin, C.-Y. Novel Liver X Receptor Ligand GAC0001E5 Disrupts Glutamine Metabolism and Induces Oxidative Stress in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9622. [Google Scholar] [CrossRef] [PubMed]
- Premaratne, A.; Ho, C.; Basu, S.; Khan, A.F.; Bawa-Khalfe, T.; Lin, C.-Y. Liver X Receptor Inverse Agonist GAC0001E5 Impedes Glutaminolysis and Disrupts Redox Homeostasis in Breast Cancer Cells. Biomolecules 2023, 13, 345. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Zheng, X.; Ma, H.; Wang, J.; Huang, M.; Fu, D.; Qin, L.; Yin, Q. Energy metabolism pathways in breast cancer progression: The reprogramming, crosstalk, and potential therapeutic targets. Transl. Oncol. 2022, 26, 101534. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yuan, L.X.; Boulbes, D.; Baek, J.M.; Wang, Y.N.; Gomez-Cabello, D.; Hawke, D.H.; Yeung, S.C.; Lee, M.H.; Hortobagyi, G.N.; et al. Fatty acid synthase phosphorylation: A novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010, 12, R96. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Foufelle, F. SREBP-1c Transcription Factor and Lipid Homeostasis: Clinical Perspective. Horm. Res. Paediatr. 2007, 68, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zou, L.; Lei, X.; Yang, X. Roles of ABCA1 in cancer. Oncol. Lett. 2022, 24, 349. [Google Scholar] [CrossRef] [PubMed]
- Wahdan-Alaswad, R.; Liu, B.; Thor, A.D. Targeted lapatinib anti-HER2/ErbB2 therapy resistance in breast cancer: Opportunities to overcome a difficult problem. Cancer Drug Resist. 2020, 3, 179. [Google Scholar] [CrossRef] [PubMed]
- Ligorio, F.; Pellegrini, I.; Castagnoli, L.; Vingiani, A.; Lobefaro, R.; Zattarin, E.; Santamaria, M.; Pupa, S.M.; Pruneri, G.; de Braud, F.; et al. Targeting lipid metabolism is an emerging strategy to enhance the efficacy of anti-HER2 therapies in HER2-positive breast cancer. Cancer Lett. 2021, 511, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Lee, M.-Y.; Park, B.-W.; Kim, K.-S. HER2 upregulates fatty acid synthase and acetyl-CoA carboxylase at a translational level in breast cancer cells. Breast Cancer Res. 2005, 7, P4.02. [Google Scholar] [CrossRef]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Med. Sci. 2000, 97, 3450–3454. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- El Ansari, R.; McIntyre, A.; Craze, M.L.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Altered glutamine metabolism in breast cancer; subtype dependencies and alternative adaptations. Histopathology 2018, 72, 183–190. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Ho, T.-S.; Ho, Y.-P.; Wong, W.-Y.; Chi-Ming Chiu, L.; Wong, Y.-S.; Eng-Choon Ooi, V. Fatty acid synthase inhibitors cerulenin and C75 retard growth and induce caspase-dependent apoptosis in human melanoma A-375 cells. Biomed. Pharmacother. 2007, 61, 578–587. [Google Scholar] [CrossRef]
- Miao, B.; Zondlo, S.; Gibbs, S.; Cromley, D.; Hosagrahara, V.P.; Kirchgessner, T.G.; Billheimer, J.; Mukherjee, R. Raising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulator. J. Lipid Res. 2004, 45, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Griffett, K.; El-Gendy, B.E.-D.M.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M.; et al. Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.J.; Valfort, A.-C.; Steinauer, N.; Chatterjee, A.; Abuirqeba, S.; Majidi, S.; Sengupta, M.; Di Paolo, R.J.; Shornick, L.P.; Zhang, J.; et al. LXR-inverse agonism stimulates immune-mediated tumor destruction by enhancing CD8 T-cell activity in triple negative breast cancer. Sci. Rep. 2019, 9, 19530. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vellon, L.; Mehmi, I.; Oza, B.P.; Ropero, S.; Colomer, R.; Lupu, R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10715–10720. [Google Scholar] [CrossRef]
- Xing, X.; Wang, S.-C.; Xia, W.; Zou, Y.; Shao, R.; Kwong, K.Y.; Yu, Z.; Zhang, S.; Miller, S.; Huang, L.; et al. The Ets protein PEA3 suppresses HER-2/neu overexpression and inhibits tumorigenesis. Nat. Med. 2000, 6, 189–195. [Google Scholar] [CrossRef]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef]
- Rais, R.; Lemberg, K.M.; Tenora, L.; Arwood, M.L.; Pal, A.; Alt, J.; Wu, Y.; Lam, J.; Aguilar, J.M.H.; Zhao, L.; et al. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Sci. Adv. 2022, 8, eabq5925. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.; Infante, J.; Arkenau, H.-T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Premaratne, A.; Basu, S.; Bagchi, A.; Zhou, T.; Feng, Q.; Lin, C.-Y. Liver X Receptor Ligand GAC0001E5 Downregulates Antioxidant Capacity and ERBB2/HER2 Expression in HER2-Positive Breast Cancer Cells. Cancers 2024, 16, 1651. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16091651
Premaratne A, Basu S, Bagchi A, Zhou T, Feng Q, Lin C-Y. Liver X Receptor Ligand GAC0001E5 Downregulates Antioxidant Capacity and ERBB2/HER2 Expression in HER2-Positive Breast Cancer Cells. Cancers. 2024; 16(9):1651. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16091651
Chicago/Turabian StylePremaratne, Asitha, Shinjini Basu, Abhinav Bagchi, Tianyi Zhou, Qin Feng, and Chin-Yo Lin. 2024. "Liver X Receptor Ligand GAC0001E5 Downregulates Antioxidant Capacity and ERBB2/HER2 Expression in HER2-Positive Breast Cancer Cells" Cancers 16, no. 9: 1651. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16091651