
Robust Enhancement of Direct Air Capture of CO2 Efficiency Using Micro-Sized Anion Exchange Resin Particles

 , and
, and
Abstract
:1. Introduction
2. Moisture-Swing Sorption Model
3. Materials and Methods
3.1. Preparation and Characterization of Amine-Based Anion Exchange Resins (IRA-900)
3.2. Absorption−Desorption Experimental
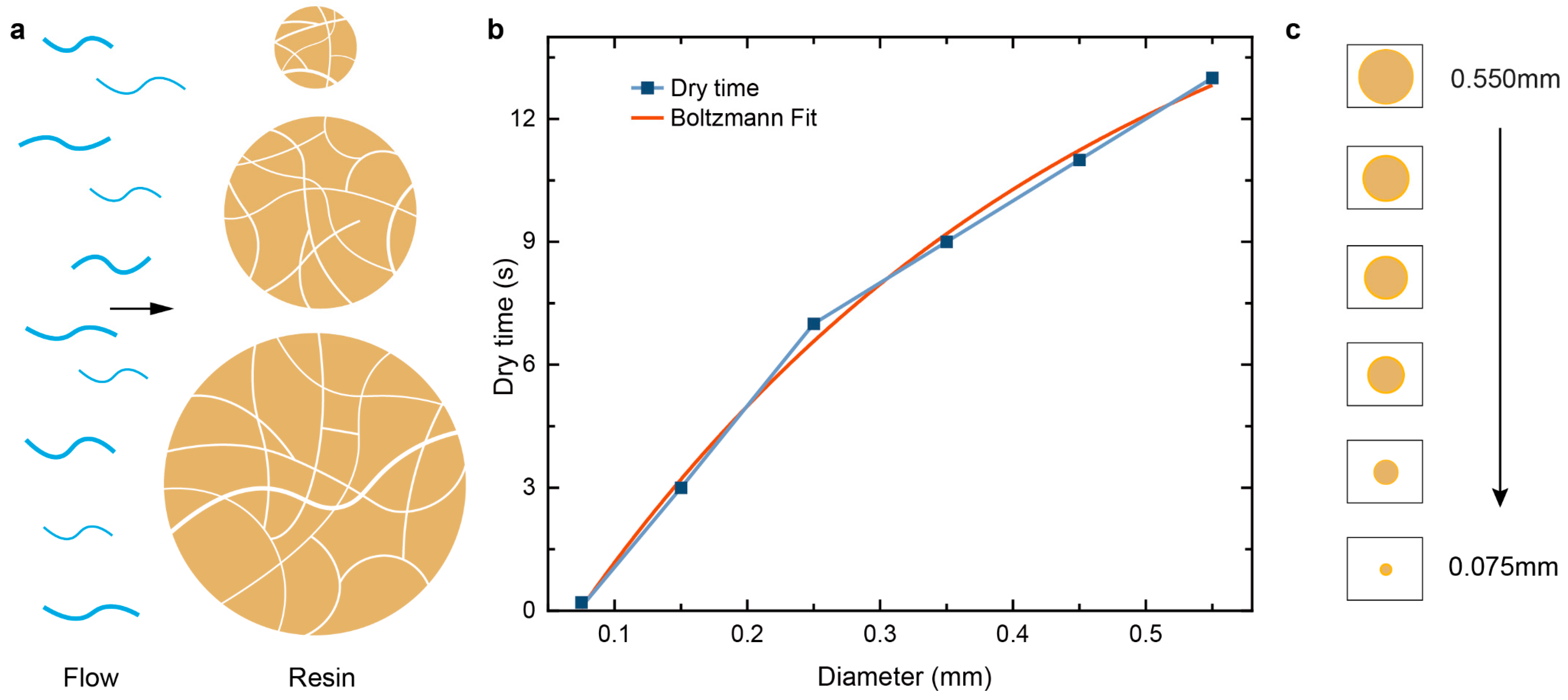
3.3. Adsorption of Water by Particles of Different Diameters
4. Results and Discussion
4.1. Adsorption and Desorption Rates of Different Diameters
4.2. Absorption Rates of Diverse Accumulations
4.3. Absorption Rates of Different Packing Densities
4.4. Multiple Tests of 0.078–0.16 mm Particles
4.5. COMSOL Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Pachauri, R.K.; Allen, M.R.; Barros, V.R.; Broome, J.; Cramer, W.; Christ, R.; Church, J.A.; Clarke, L.; Dahe, Q.; Dasgupta, P. Climate change 2014: Synthesis report. In Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2014. [Google Scholar]
- Wang, X.; Song, J.; Chen, Y.; Xiao, H.; Shi, X.; Liu, Y.; Zhu, L.; He, Y.-L.; Chen, X. CO2 absorption over ion exchange resins: The effect of amine functional groups and microporous structures. Ind. Eng. Chem. Res. 2020, 59, 16507–16515. [Google Scholar] [CrossRef]
- Xu, J.; Shi, X.; Li, Q.; Wang, T.; Lackner, K.S. Kinetic analysis of an anion exchange absorbent for CO2 capture from ambient air. PLoS ONE 2017, 12, e0179828. [Google Scholar] [CrossRef]
- Solomon, S. Climate Change 2007-the Physical Science Basis: Working Group I Contribution to the Fourth Assessment Report of the IPCC; Cambridge University Press: Cambridge, UK, 2007; Volume 4. [Google Scholar]
- Lackner, K.S.; Brennan, S.; Matter, J.M.; Park, A.-H.A.; Wright, A.; Van Der Zwaan, B. The urgency of the development of CO2 capture from ambient air. Proc. Natl. Acad. Sci. USA 2012, 109, 13156–13162. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, L.; Wang, J.; Cabot, A.; Zhu, Y. Boosting Hydrogen Evolution by Methanol Oxidation Reaction on Ni-Based Electrocatalysts: From Fundamental Electrochemistry to Perspectives. ACS Energy Lett. 2024, 9, 853–879. [Google Scholar] [CrossRef]
- Keith, D.W. Why capture CO2 from the atmosphere? Science 2009, 325, 1654–1655. [Google Scholar] [CrossRef] [PubMed]
- Lackner, K.S. A guide to CO2 sequestration. Science 2003, 300, 1677–1678. [Google Scholar] [CrossRef]
- Lackner, K.S.; Aines, R.; Atkins, S.; AtKisson, A.; Barrett, S.; Barteau, M.; Braun, R.J.; Brouwer, J.; Broecker, W.; Browne, J.B. The promise of negative emissions. Science 2016, 354, 714. [Google Scholar] [CrossRef] [PubMed]
- Scott, V.; Gilfillan, S.; Markusson, N.; Chalmers, H.; Haszeldine, R.S. Last chance for carbon capture and storage. Nat. Clim. Chang. 2013, 3, 105–111. [Google Scholar] [CrossRef]
- Lackner, K.S. Capture of carbon dioxide from ambient air. Eur. Phys. J. Spec. Top. 2009, 176, 93–106. [Google Scholar] [CrossRef]
- Shi, X.; Xiao, H.; Azarabadi, H.; Song, J.; Wu, X.; Chen, X.; Lackner, K.S. Sorbents for the direct capture of CO2 from ambient air. Angew. Chem. Int. Ed. 2020, 59, 6984–7006. [Google Scholar] [CrossRef]
- Shi, X.; Lee, G.A.; Liu, S.; Kim, D.; Alahmed, A.; Jamal, A.; Wang, L.; Park, A.-H.A. Water-stable MOFs and hydrophobically encapsulated MOFs for CO2 capture from ambient air and wet flue gas. Mater. Today 2023, 65, 207–226. [Google Scholar] [CrossRef]
- Shi, X.; Lin, Y.; Chen, X. Development of sorbent materials for direct air capture of CO2. MRS Bull. 2022, 47, 405–415. [Google Scholar] [CrossRef]
- Shu, Q.; Haug, M.; Tedesco, M.; Kuntke, P.; Hamelers, H.V. Direct air capture using electrochemically regenerated anion exchange resins. Environ. Sci. Technol. 2022, 56, 11559–11566. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; De, M. Room temperature adsorptive removal of thiophene over zinc oxide-based adsorbents. J. Mater. Eng. Perform. 2018, 27, 2661–2667. [Google Scholar] [CrossRef]
- Sanz-Pérez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct capture of CO2 from ambient air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; De, M. Carbon Dioxide Removal by Chemically and Thermally Reduced Graphene-Based Adsorbents. Korean J. Chem. Eng. 2024, 41, 783–796. [Google Scholar] [CrossRef]
- Wang, T.; Lackner, K.S.; Wright, A. Moisture Swing Sorbent for Carbon Dioxide Capture from Ambient Air. Environ. Sci. Technol. 2011, 45, 6670–6675. [Google Scholar] [CrossRef]
- Shi, X.; Xiao, H.; Lackner, K.S.; Chen, X. Capture CO2 from Ambient Air Using Nanoconfined Ion Hydration. Angew. Chem. 2016, 128, 4094–4097. [Google Scholar] [CrossRef]
- Song, J.; Zhu, L.; Shi, X.; Liu, Y.; Xiao, H.; Chen, X. Moisture swing ion-exchange resin-PO4 sorbent for reversible CO2 capture from ambient air. Energy Fuels 2019, 33, 6562–6567. [Google Scholar] [CrossRef]
- Shi, X.; Xiao, H.; Kanamori, K.; Yonezu, A.; Lackner, K.S.; Chen, X. Moisture-Driven CO2 Sorbents. Joule 2020, 4, 1823–1837. [Google Scholar] [CrossRef]
- Lackner, K. Thermodynamics of the humidity swing driven air capture of carbon dioxide. GRT LLC Tucson AZ 2008. [Google Scholar]
- Elimelech, M.; Phillip, W.A. The future of seawater desalination: Energy, technology, and the environment. Science 2011, 333, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Gude, V.G. Desalination and sustainability–an appraisal and current perspective. Water Res. 2016, 89, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, P.; Tobias, D.J. Specific ion effects at the air/water interface. Chem. Rev. 2006, 106, 1259–1281. [Google Scholar] [CrossRef] [PubMed]
- Lackner, K.S. The thermodynamics of direct air capture of carbon dioxide. Energy 2013, 50, 38–46. [Google Scholar] [CrossRef]
- Lawlor, D.W.; Cornic, G. Photosynthetic carbon assimilation and associated metabolism in relation to water deficits in higher plants. Plant Cell Environ. 2002, 25, 275–294. [Google Scholar] [CrossRef]
- Chaves, M. Effects of water deficits on carbon assimilation. J. Exp. Bot. 1991, 42, 1–16. [Google Scholar] [CrossRef]
- Socolow, R.; Desmond, M.; Aines, R.; Blackstock, J.; Bolland, O.; Kaarsberg, T.; Lewis, N.; Mazzotti, M.; Pfeffer, A.; Sawyer, K. Direct Air Capture of CO2 with Chemicals: A Technology Assessment for the APS Panel on Public Affairs; American Physical Society: Washington, DC, USA, 2011. [Google Scholar]
- Wang, T.; Lackner, K.S.; Wright, A.B. Moisture-swing sorption for carbon dioxide capture from ambient air: A thermodynamic analysis. Phys. Chem. Chem. Phys. 2013, 15, 504–514. [Google Scholar] [CrossRef]
- Shi, X.; Xiao, H.; Liao, X.; Armstrong, M.; Chen, X.; Lackner, K.S. Humidity effect on ion behaviors of moisture-driven CO2 sorbents. J. Chem. Phys. 2018, 149, 164708. [Google Scholar] [CrossRef]
- McDonald, T.M.; Lee, W.R.; Mason, J.A.; Wiers, B.M.; Hong, C.S.; Long, J.R. Capture of carbon dioxide from air and flue gas in the alkylamine-appended metal–organic framework mmen-Mg2 (dobpdc). J. Am. Chem. Soc. 2012, 134, 7056–7065. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; May, R.B.; Prakash, G.S.; Olah, G.A.; Narayanan, S. Carbon dioxide capture from the air using a polyamine based regenerable solid adsorbent. J. Am. Chem. Soc. 2011, 133, 20164–20167. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Kang, D.Y.; Copeland, J.R.; Bollini, P.; Sievers, C.; Kamegawa, T.; Yamashita, H.; Jones, C.W. Enhanced CO2 adsorption over polymeric amines supported on heteroatom-incorporated SBA-15 silica: Impact of heteroatom type and loading on sorbent structure and adsorption performance. Chem. A Eur. J. 2012, 18, 16649–16664. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Kang, D.-Y.; Copeland, J.R.; Brunelli, N.A.; Didas, S.A.; Bollini, P.; Sievers, C.; Kamegawa, T.; Yamashita, H.; Jones, C.W. Dramatic enhancement of CO2 uptake by poly (ethyleneimine) using zirconosilicate supports. J. Am. Chem. Soc. 2012, 134, 10757–10760. [Google Scholar] [CrossRef]
- Pang, S.H.; Lively, R.P.; Jones, C.W. Oxidatively-stable linear poly (propylenimine)-containing adsorbents for CO2 capture from ultradilute streams. ChemSusChem 2018, 11, 2628–2637. [Google Scholar] [CrossRef]
- Kumar, A.; Madden, D.G.; Lusi, M.; Chen, K.J.; Daniels, E.A.; Curtin, T.; Perry IV, J.J.; Zaworotko, M.J. Direct air capture of CO2 by physisorbent materials. Angew. Chem. Int. Ed. 2015, 54, 14372–14377. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.H.; Lee, L.-C.; Sakwa-Novak, M.A.; Lively, R.P.; Jones, C.W. Design of aminopolymer structure to enhance performance and stability of CO2 sorbents: Poly (propylenimine) vs poly (ethylenimine). J. Am. Chem. Soc. 2017, 139, 3627–3630. [Google Scholar] [CrossRef]
- Lin, R.-B.; Xiang, S.; Zhou, W.; Chen, B. Microporous metal-organic framework materials for gas separation. Chem 2020, 6, 337–363. [Google Scholar] [CrossRef]
- Madden, D.G.; Scott, H.S.; Kumar, A.; Chen, K.-J.; Sanii, R.; Bajpai, A.; Lusi, M.; Curtin, T.; Perry, J.J.; Zaworotko, M.J. Flue-gas and direct-air capture of CO2 by porous metal–organic materials. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2017, 375, 20160025. [Google Scholar] [CrossRef]
- Keller, L.; Ohs, B.; Lenhart, J.; Abduly, L.; Blanke, P.; Wessling, M. High capacity polyethylenimine impregnated microtubes made of carbon nanotubes for CO2 capture. Carbon 2018, 126, 338–345. [Google Scholar] [CrossRef]
- Sayari, A.; Belmabkhout, Y. Stabilization of amine-containing CO2 adsorbents: Dramatic effect of water vapor. J. Am. Chem. Soc. 2010, 132, 6312–6314. [Google Scholar] [CrossRef]
- Li, P.; Liu, W.; Dennis, J.S.; Zeng, H.C. Synthetic architecture of MgO/C nanocomposite from hierarchical-structured coordination polymer toward enhanced CO2 capture. ACS Appl. Mater. Interfaces 2017, 9, 9592–9602. [Google Scholar] [CrossRef] [PubMed]
- Sepahvand, S.; Jonoobi, M.; Ashori, A.; Gauvin, F.; Brouwers, H.; Oksman, K.; Yu, Q. A promising process to modify cellulose nanofibers for carbon dioxide (CO2) adsorption. Carbohydr. Polym. 2020, 230, 115571. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sorbent | Adsorption | Capacity (mmol/g) | Capacity Stability | Ref. |
|---|---|---|---|---|
| IRA-900 (0.074–0.160 mm) | 500 ppm 25 °C 1 atm | 0.58 | 100 cycles, 100% | / |
| Mg-MOF-74 | 400 ppm 25 °C 1 atm | 0.14 | 180 °C desorption, stable | [33] |
| FS-PEI-33 | 400 ppm 23 °C | 1.18 | 4 cycles, 96% | [34] |
| Zr-SBA-15 | 400 ppm 25 °C 1 atm | 0.85 | 4 cycles, 98% | [35,36] |
| SBA-15 | 400 ppm 35 °C | 0.76 | 50 cycles, stable, 110 °C desorption | [37] |
| HKUST-1 | 400 ppm 25 °C 1 atm | 0.05 | 140 °C desorption, stable | [38] |
| PPI/Porous Silica | 400 ppm 35 °C | 0.20–3.60 | 4 cycles, 88% | [39] |
| Diamine/MOF | 25 °C 1 atm | 1.55 | 5 cycles, stable | [40] |
| SIFSIX-3-Ni | 400 ppm 25 °C 1 atm | 0.18 | 140 °C desorption, stable | [41] |
| Carbon Nanotube | 30 °C 1 atm | 1.07 | 10 cycles, 90% | [42] |
| PE-MCM-41 | (−5 °C)–(+5 °C) RH 60–80% | 0.40 | 3 cycles, 4% | [43] |
| NaOH | 380 ppm 1 atm | / | 900 °C regeneration, stable | [26] |
| 20 wt% MgO-RHA | 26 °C 10% CO2/N2 1 atm | 4.56 | 10 cycles, 92% | [44] |
| Poly HIPE/nano-TiO2/PEI-50 | 75 °C CO2/H2O/N2 (1:1:8) | 5.59 | 50 cycles, 91% | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Hu, J.; Zhang, F.; Zhu, J.; Shi, X.; Wang, L. Robust Enhancement of Direct Air Capture of CO2 Efficiency Using Micro-Sized Anion Exchange Resin Particles. Sustainability 2024, 16, 3601. https://0-doi-org.brum.beds.ac.uk/10.3390/su16093601
Liu S, Hu J, Zhang F, Zhu J, Shi X, Wang L. Robust Enhancement of Direct Air Capture of CO2 Efficiency Using Micro-Sized Anion Exchange Resin Particles. Sustainability. 2024; 16(9):3601. https://0-doi-org.brum.beds.ac.uk/10.3390/su16093601
Chicago/Turabian StyleLiu, Shuohan, Junqiang Hu, Fan Zhang, Jianzhong Zhu, Xiaoyang Shi, and Lei Wang. 2024. "Robust Enhancement of Direct Air Capture of CO2 Efficiency Using Micro-Sized Anion Exchange Resin Particles" Sustainability 16, no. 9: 3601. https://0-doi-org.brum.beds.ac.uk/10.3390/su16093601