

Darunavir Nanoformulation Suppresses HIV Pathogenesis in Macrophages and Improves Drug Delivery to the Brain in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PLGA-DRV
2.3. Characterization of PLGA Nanoparticles
2.4. Encapsulation Efficiency and Drug Loading
2.5. DRV Quantification by LC-MS/MS
2.6. In Vitro Drug Release
2.7. Cell Culturing
2.8. LDH Cell Viability Assay
2.9. HIV Type 1 p24 ELISA
2.10. Total Antioxidant Capacity (TAC)
2.11. Quantification of Intracellular Reactive Oxygen Species (ROS) with Fluorescence-Based Assay
2.12. Detection of Cytokines and Chemokines
2.13. Animal Studies
3. Results
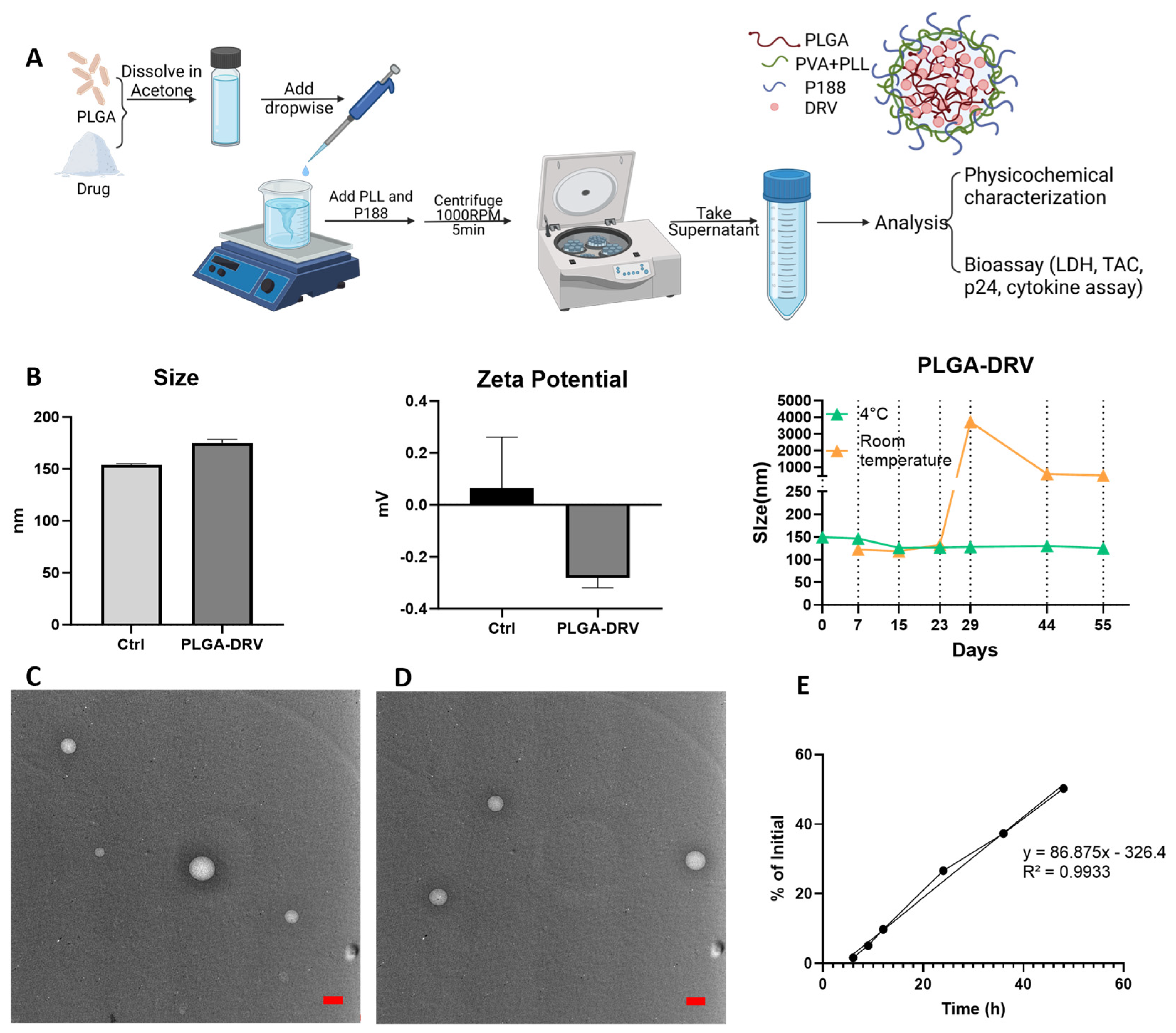
3.1. Characterization of PLGA-DRV
3.2. Effect of PLGA NPs on Cytotoxicity in U1 Macrophages in the Absence and Presence of In Vitro BBB Model
3.3. Effect of PLGA NPs on Antioxidant Capacity and Oxidative Stress in U1 Macrophages in the Absence and Presence of In Vitro BBB Model
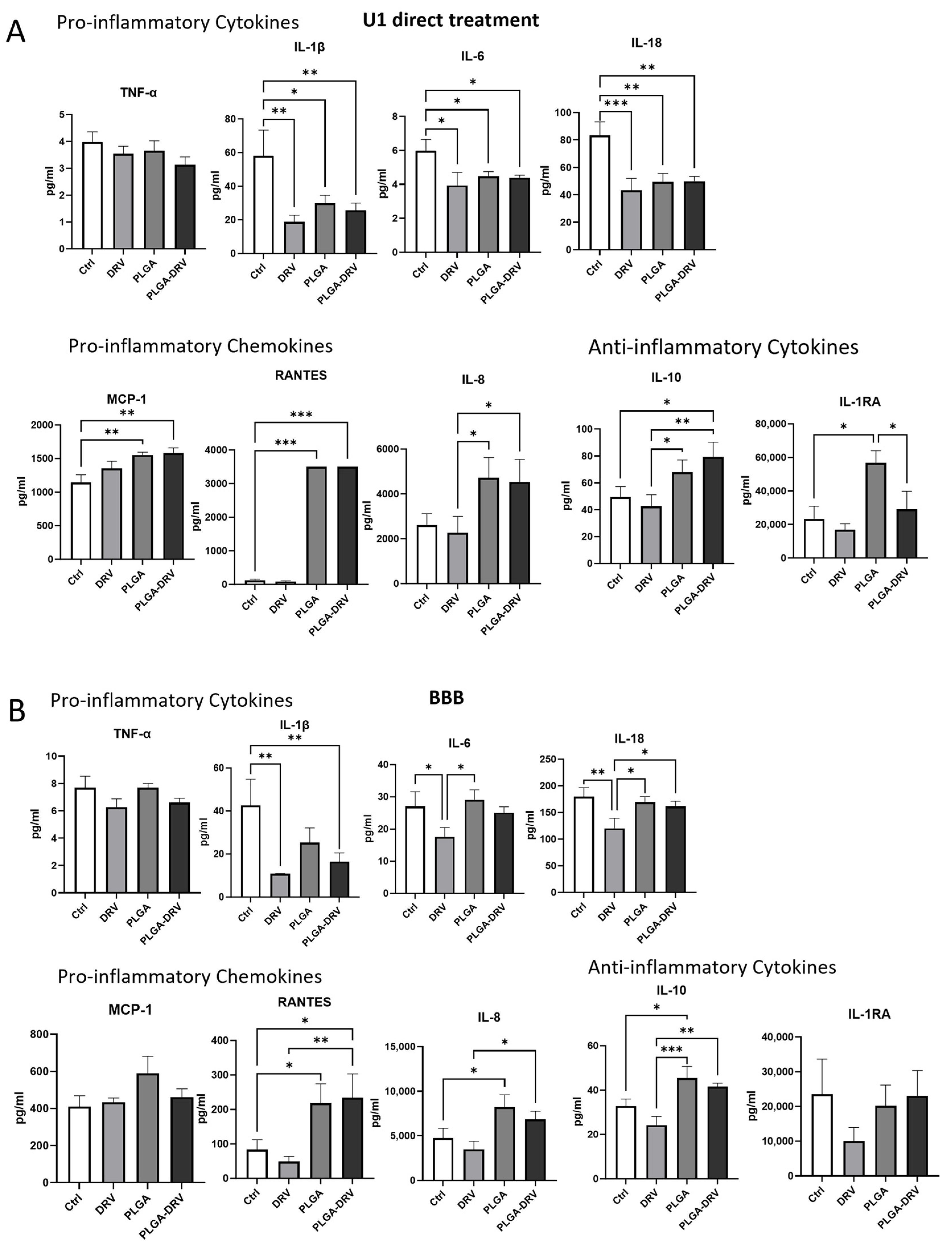
3.4. Effect of PLGA NPs on Cytokines and Chemokines in U1 Macrophages in the Absence and Presence of In Vitro BBB Model
3.5. Intracellular DRV Concentration
3.6. Effect of PLGA NPs in Viral Suppression in U1 Macrophages in the Absence and Presence of In Vitro BBB
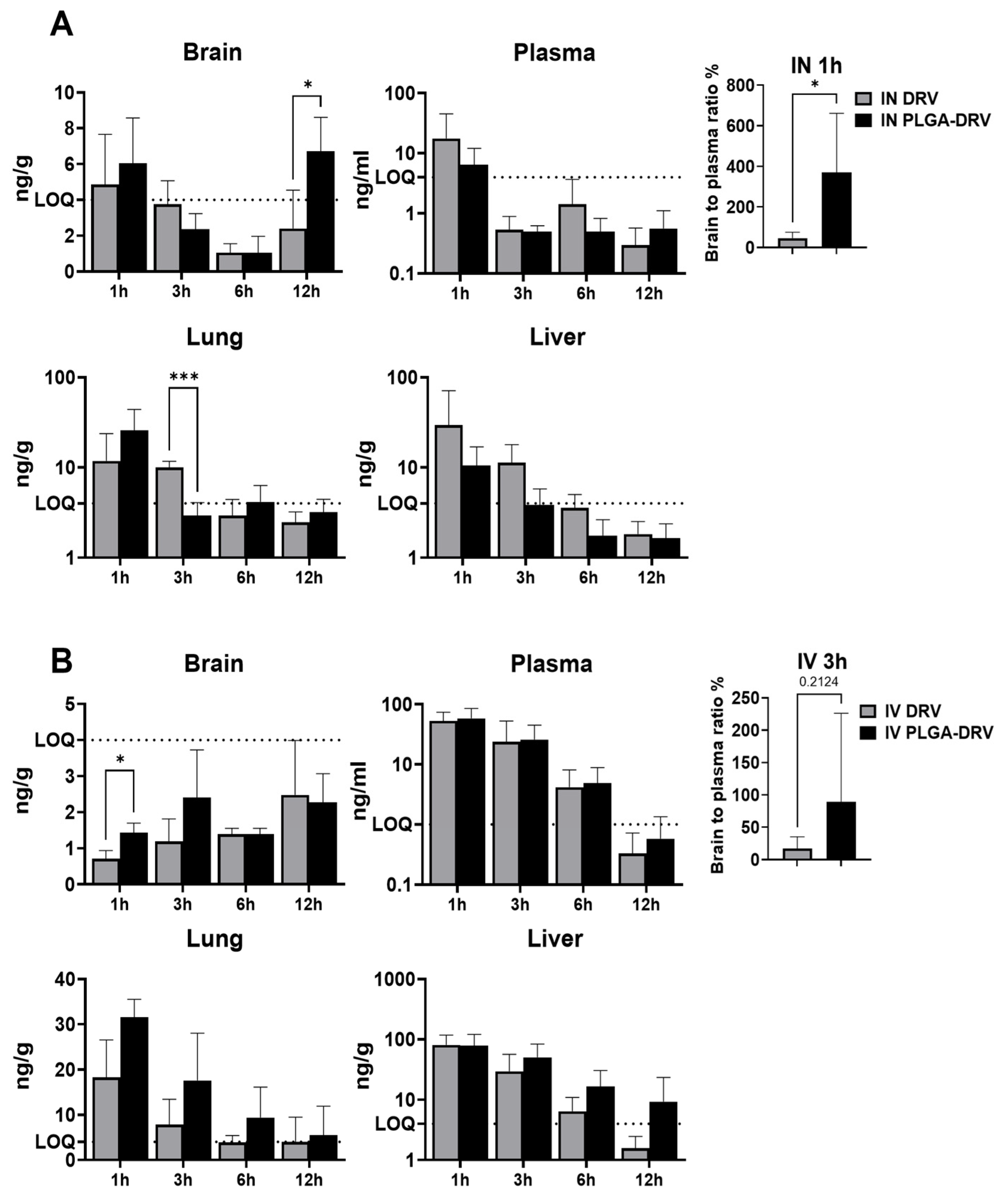
3.7. DRV Levels in Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valcour, V.; Chalermchai, T.; Sailasuta, N.; Marovich, M.; Lerdlum, S.; Suttichom, D.; Suwanwela, N.C.; Jagodzinski, L.; Michael, N.; Spudich, S.; et al. Central nervous system viral invasion and inflammation during acute HIV infection. J. Infect. Dis. 2012, 206, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, A.; Spuch, C.; Penedo, M.A.; Olivares, J.M.; Agís-Balboa, R.C. Important role of microglia in HIV-1 associated neurocognitive disorders and the molecular pathways implicated in its pathogenesis. Ann. Med. 2021, 53, 43–69. [Google Scholar] [CrossRef] [PubMed]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Al-Harti, L.; Joseph, J.; Nath, A. Astrocytes as an HIV CNS reservoir: Highlights and reflections of an NIMH-sponsored symposium. J. Neurovirol. 2018, 24, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Colosi, D.A.; Rao, V.R. HIV-1 Induced CNS Dysfunction: Current Overview and Research Priorities. Curr. HIV Res. 2016, 14, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Haas, D.W.; Johnson, B.; Nicotera, J.; Bailey, V.L.; Harris, V.L.; Bowles, F.B.; Raffanti, S.; Schranz, J.; Finn, T.S.; Saah, A.J.; et al. Effects of ritonavir on indinavir pharmacokinetics in cerebrospinal fluid and plasma. Antimicrob. Agents Chemother. 2003, 47, 2131–2137. [Google Scholar] [CrossRef]
- Decloedt, E.H.; Rosenkranz, B.; Maartens, G.; Joska, J. Central nervous system penetration of antiretroviral drugs: Pharmacokinetic, pharmacodynamic and pharmacogenomic considerations. Clin. Pharmacokinet. 2015, 54, 581–598. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Luissint, A.-C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005, 2, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Gonzalez-Martinez, J.; Hossain, M.; Cucullo, L.; Fazio, V.; Janigro, D.; Marchi, N. Pattern of P450 expression at the human blood-brain barrier: Roles of epileptic condition and laminar flow. Epilepsia 2010, 51, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.; Jayant, R.D.; Kaushik, A.; Sagar, V. Getting into the brain: Potential of nanotechnology in the management of NeuroAIDS. Adv. Drug Deliv. Rev. 2016, 103, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood-Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43, 695–708. [Google Scholar] [CrossRef]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Letendre, S.L.; FCEllis, R.J.; Clifford, D.; Collier, A.C.; Gelman, B.; Marra, C.; McArthur, J.; McCutchan, J.A.; Morgello, S.; Simpson, D.; et al. CNS Penetration Effectiveness (CPE) Ranks. In Proceedings of the 17th Conference on Retroviruses and Opportunistic Infections, San Francisco, CA, USA, 16–19 February 2010. [Google Scholar]
- Mukerji, S.S.; Misra, V.; Lorenz, D.R.; Uno, H.; Morgello, S.; Franklin, D.; Ellis, R.J.; Letendre, S.; Gabuzda, D. Impact of Antiretroviral Regimens on Cerebrospinal Fluid Viral Escape in a Prospective Multicohort Study of Antiretroviral Therapy-Experienced Human Immunodeficiency Virus-1-Infected Adults in the United States. Clin. Infect. Dis. 2018, 67, 1182–1190. [Google Scholar] [CrossRef]
- Gong, Y.; Chowdhury, P.; Nagesh, P.K.B.; Rahman, M.A.; Zhi, K.; Yallapu, M.M.; Kumar, S. Novel elvitegravir nanoformulation for drug delivery across the blood-brain barrier to achieve HIV-1 suppression in the CNS macrophages. Sci. Rep. 2020, 10, 3835. [Google Scholar] [CrossRef]
- Gong, Y.; Zhi, K.; Nagesh, P.K.B.; Sinha, N.; Chowdhury, P.; Chen, H.; Gorantla, S.; Yallapu, M.M.; Kumar, S. An Elvitegravir Nanoformulation Crosses the Blood–Brain Barrier and Suppresses HIV-1 Replication in Microglia. Viruses 2020, 12, 564. [Google Scholar] [CrossRef]
- Pinto, M.; Silva, V.; Barreiro, S.; Silva, R.; Remião, F.; Borges, F.; Fernandes, C. Brain drug delivery and neurodegenerative diseases: Polymeric PLGA-based nanoparticles as a forefront platform. Ageing Res. Rev. 2022, 79, 101658. [Google Scholar] [CrossRef]
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Palocci, C.; Valletta, A.; Chronopoulou, L.; Donati, L.; Bramosanti, M.; Brasili, E.; Baldan, B.; Pasqua, G. Endocytic pathways involved in PLGA nanoparticle uptake by grapevine cells and role of cell wall and membrane in size selection. Plant Cell Rep. 2017, 36, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chowdhury, P.; Midde, N.M.; Rahman, M.A.; Yallapu, M.M.; Kumar, S. Novel elvitegravir nanoformulation approach to suppress the viral load in HIV-infected macrophages. Biochem. Biophys. Rep. 2017, 12, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Bartels, H.; Decosterd, L.; Battegay, M.; Marzolini, C. Darunavir concentrations in CSF of HIV-infected individuals when boosted with cobicistat versus ritonavir. J. Antimicrob. Chemother. 2017, 72, 2574–2577. [Google Scholar] [CrossRef] [PubMed]
- Letendre, S.L.; Ellis, R.J.; Ances, B.M.; McCutchan, J.A. Neurologic complications of HIV disease and their treatment. Top. HIV Med. 2010, 18, 45–55. [Google Scholar] [PubMed]
- Yallapu, M.M.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J. Colloid. Interface Sci. 2010, 351, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Midde, N.M.; Gong, Y.; Cory, T.J.; Li, J.; Meibohm, B.; Li, W.; Kumar, S. Influence of Ethanol on Darunavir Hepatic Clearance and Intracellular PK/PD in HIV-Infected Monocytes, and CYP3A4-Darunavir Interactions Using Inhibition and in Silico Binding Studies. Pharm. Res. 2017, 34, 1925–1933. [Google Scholar] [CrossRef]
- Kumar, A.; Zhou, L.; Godse, S.; Sinha, N.; Ma, D.; Parmar, K.; Kumar, S. Intranasal delivery of darunavir improves brain drug concentrations in mice for effective HIV treatment. Biochem. Biophys. Rep. 2023, 33, 101408. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow. Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Shi, D.; Mi, G.; Bhattacharya, S.; Nayar, S.; Webster, T.J. Optimizing superparamagnetic iron oxide nanoparticles as drug carriers using an in vitro blood-brain barrier model. Int. J. Nanomed. 2016, 11, 5371–5379. [Google Scholar] [CrossRef]
- Kumar, A.; Sinha, N.; Kodidela, S.; Zhou, L.; Singh, U.P.; Kumar, S. Effect of benzo(a)pyrene on oxidative stress and inflammatory mediators in astrocytes and HIV-infected macrophages. PLoS ONE 2022, 17, e0275874. [Google Scholar] [CrossRef] [PubMed]
- Godse, S.; Zhou, L.; Sinha, N.; Kodidela, S.; Kumar, A.; Singh, U.P.; Kumar, S. Curcumin enhances elvitegravir concentration and alleviates oxidative stress and inflammatory response. Sci. Rep. 2023, 13, 19864. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Kruize, Z.; Kootstra, N.A. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front. Microbiol. 2019, 10, 2828. [Google Scholar] [CrossRef] [PubMed]
- Boonyagars, L.; Kiatsoongsong, N.; Winitprichagul, S. HIV-Associated Dementia: Associated Factors and Characteristics of Cognitive Domain Abnormalities in Elderly People Living with HIV Treated with Highly Active Antiretroviral Therapy. Am. J. Trop. Med. Hyg. 2022, 107, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Kodidela, S.; Godse, S.; Thomas-Gooch, S.; Kumar, A.; Raji, B.; Zhi, K.; Kochat, H.; Kumar, S. Targeted Drug Delivery to the Central Nervous System Using Extracellular Vesicles. Pharmaceuticals 2022, 15, 358. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Izadkhashti, A.; Price, R.W.; Mallon, P.W.; De Meulder, M.; Timmerman, P.; Gisslén, M. Darunavir concentrations in cerebrospinal fluid and blood in HIV-1-infected individuals. AIDS Res. Hum. Retroviruses 2009, 25, 457–461. [Google Scholar] [CrossRef]
- Alford, K.; Banerjee, S.; Nixon, E.; O’Brien, C.; Pounds, O.; Butler, A.; Elphick, C.; Henshaw, P.; Anderson, S.; Vera, J.H. Assessment and Management of HIV-Associated Cognitive Impairment: Experience from a Multidisciplinary Memory Service for People Living with HIV. Brain Sci. 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Latronico, T.; Rizzi, F.; Panniello, A.; Laquintana, V.; Arduino, I.; Denora, N.; Fanizza, E.; Milella, S.; Mastroianni, C.M.; Striccoli, M.; et al. Luminescent PLGA Nanoparticles for Delivery of Darunavir to the Brain and Inhibition of Matrix Metalloproteinase-9, a Relevant Therapeutic Target of HIV-Associated Neurological Disorders. ACS Chem. Neurosci. 2021, 12, 4286–4301. [Google Scholar] [CrossRef]
- Yan, J.; Huang, L.; Feng, J.; Yang, X. The Recent Applications of PLGA-Based Nanostructures for Ischemic Stroke. Pharmaceutics 2023, 15, 2322. [Google Scholar] [CrossRef]
- Anand, B.; Wu, Q.; Nakhaei-Nejad, M.; Karthivashan, G.; Dorosh, L.; Amidian, S.; Dahal, A.; Li, X.; Stepanova, M.; Wille, H.; et al. Significance of native PLGA nanoparticles in the treatment of Alzheimer’s disease pathology. Bioact. Mater. 2022, 17, 506–525. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, M.; Zeng, F.; Jin, H.; Xu, Q.; Huang, Y. Dual-Targeting Magnetic PLGA Nanoparticles for Codelivery of Paclitaxel and Curcumin for Brain Tumor Therapy. ACS Appl. Mater. Interfaces 2016, 8, 32159–32169. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tong, Y.; Bai, L.; Ye, L.; Zhong, L.; Duan, X.; Zhu, Y. Lactoferrin functionalized PEG-PLGA nanoparticles of shikonin for brain targeting therapy of glioma. Int. J. Biol. Macromol. 2018, 107, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Gambaryan, P.Y.; Kondrasheva, I.G.; Severin, E.S.; Guseva, A.A.; Kamensky, A.A. Increasing the Efficiency of Parkinson’s Disease Treatment Using a poly(lactic-co-glycolic acid) (PLGA) Based L-DOPA Delivery System. Exp. Neurobiol. 2014, 23, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Monge-Fuentes, V.; Biolchi Mayer, A.; Lima, M.R.; Geraldes, L.R.; Zanotto, L.N.; Moreira, K.G.; Martins, O.P.; Piva, H.L.; Felipe, M.S.S.; Amaral, A.C.; et al. Dopamine-loaded nanoparticle systems circumvent the blood–brain barrier restoring motor function in mouse model for Parkinson’s Disease. Sci. Rep. 2021, 11, 15185. [Google Scholar] [CrossRef] [PubMed]
- Hanada, S.; Fujioka, K.; Inoue, Y.; Kanaya, F.; Manome, Y.; Yamamoto, K. Cell-based in vitro blood-brain barrier model can rapidly evaluate nanoparticles’ brain permeability in association with particle size and surface modification. Int. J. Mol. Sci. 2014, 15, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Macri, L.; Kohn, J. Formulation Strategy for the Delivery of Cyclosporine A: Comparison of Two Polymeric Nanospheres. Sci. Rep. 2015, 5, 13065. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.M.; do Nascimento, T.C.F.; Casa, D.M.; Dalmolin, L.F.; de Mattos, A.C.; Hoss, I.; Romano, M.A.; Mainardes, R.M. Pharmacokinetics of curcumin-loaded PLGA and PLGA–PEG blend nanoparticles after oral administration in rats. Colloids Surf. B Biointerfaces 2013, 101, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.L.; Bajaj, P.; Whitehead, K.A. Achieving long-term stability of lipid nanoparticles: Examining the effect of pH, temperature, and lyophilization. Int. J. Nanomed. 2017, 12, 305–315. [Google Scholar] [CrossRef]
- Fonte, P.; Soares, S.; Costa, A.; Andrade, J.C.; Seabra, V.; Reis, S.; Sarmento, B. Effect of cryoprotectants on the porosity and stability of insulin-loaded PLGA nanoparticles after freeze-drying. Biomatter 2012, 2, 329–339. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, S.M.; Chen, P.P.; Cheng, L.; Zhou, W.; Tang, W.X.; Chen, Z.W.; Ke, C.M. Controlled release of insulin from PLGA nanoparticles embedded within PVA hydrogels. J. Mater. Sci. Mater. Med. 2007, 18, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxidative Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.; Nelson, B.; Oputiri, D.; Geoffrey, O.-B.P. Antiretroviral Toxicity and Oxidative Stress. Am. J. Pharmacol. Toxicol. 2013, 8, 187. [Google Scholar] [CrossRef]
- Mandas, A.; Iorio, E.L.; Congiu, M.G.; Balestrieri, C.; Mereu, A.; Cau, D.; Dessì, S.; Curreli, N. Oxidative imbalance in HIV-1 infected patients treated with antiretroviral therapy. J. Biomed. Biotechnol. 2009, 2009, 749575. [Google Scholar] [CrossRef] [PubMed]
- Opii, W.O.; Sultana, R.; Abdul, H.M.; Ansari, M.A.; Nath, A.; Butterfield, D.A. Oxidative stress and toxicity induced by the nucleoside reverse transcriptase inhibitor (NRTI)—2′,3′-dideoxycytidine (ddC): Relevance to HIV-dementia. Exp. Neurol. 2007, 204, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; Crowe, S.M. Cytokines and HIV-1: Interactions and clinical implications. Antivir. Chem. Chemother. 2001, 12, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Spudich, S.; González-Scarano, F. HIV-1-related central nervous system disease: Current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb. Perspect. Med. 2012, 2, a007120. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Eugenin, E.A. Mechanisms of HIV Neuropathogenesis: Role of Cellular Communication Systems. Curr. HIV Res. 2016, 14, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Mazzuti, L.; Turriziani, O.; Mezzaroma, I. The Many Faces of Immune Activation in HIV-1 Infection: A Multifactorial Interconnection. Biomedicines 2023, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Osuji, F.N.; Onyenekwe, C.C.; Ahaneku, J.E.; Ukibe, N.R. The effects of highly active antiretroviral therapy on the serum levels of pro-inflammatory and anti-inflammatory cytokines in HIV infected subjects. J. Biomed. Sci. 2018, 25, 88. [Google Scholar] [CrossRef]
- Muema, D.M.; Akilimali, N.A.; Ndumnego, O.C.; Rasehlo, S.S.; Durgiah, R.; Ojwach, D.B.A.; Ismail, N.; Dong, M.; Moodley, A.; Dong, K.L.; et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Med. 2020, 18, 81. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.R.; Markovitz, D.M.; Woodford, N.L.; Rochford, R.; Strieter, R.M.; Coffey, M.J. TNF-alpha inhibits HIV-1 replication in peripheral blood monocytes and alveolar macrophages by inducing the production of RANTES and decreasing C-C chemokine receptor 5 (CCR5) expression. J. Immunol. 1999, 163, 3653–3661. [Google Scholar] [CrossRef]
- Trkola, A.; Paxton, W.A.; Monard, S.P.; Hoxie, J.A.; Siani, M.A.; Thompson, D.A.; Wu, L.; Mackay, C.R.; Horuk, R.; Moore, J.P. Genetic Subtype-Independent Inhibition of Human Immunodeficiency Virus Type 1 Replication by CC and CXC Chemokines. J. Virol. 1998, 72, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, F.; DeVico, A.L.; Garzino-Demo, A.; Arya, S.K.; Gallo, R.C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar] [CrossRef]
- Kodidela, S.; Sinha, N.; Kumar, A.; Zhou, L.; Godse, S.; Kumar, S. Extracellular vesicles released from macrophages modulates interleukin-1β in astrocytic and neuronal cells. Sci. Rep. 2023, 13, 3005. [Google Scholar] [CrossRef]
- Borges, Á.H.; O’Connor, J.L.; Phillips, A.N.; Rönsholt, F.F.; Pett, S.; Vjecha, M.J.; French, M.A.; Lundgren, J.D.; Insight Smart; Esprit Study Groups; et al. Factors Associated with Plasma IL-6 Levels During HIV Infection. J. Infect. Dis. 2015, 212, 585–595. [Google Scholar] [CrossRef]
- Borges, Á.H.; O’Connor, J.L.; Phillips, A.N.; Neaton, J.D.; Grund, B.; Neuhaus, J.; Vjecha, M.J.; Calmy, A.; Koelsch, K.K.; Lundgren, J.D. Interleukin 6 Is a Stronger Predictor of Clinical Events Than High-Sensitivity C-Reactive Protein or D-Dimer During HIV Infection. J. Infect. Dis. 2016, 214, 408–416. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Valverde-Villegas, J.M.; Kaminski, V.d.L.; de Medeiros, R.M.; Almeida, S.E.d.M.; Santos, B.R.; de Melo, M.G.; Hackenhaar, F.S.; Chies, J.A.B. Increased IL-8 levels in HIV-infected individuals who initiated ART with CD4+ T cell counts <350 cells/mm3—A potential hallmark of chronic inflammation. Microbes Infect. 2020, 22, 474–480. [Google Scholar]
- Feria, M.G.; Taborda, N.A.; Hernandez, J.C.; Rugeles, M.T. HIV replication is associated to inflammasomes activation, IL-1β, IL-18 and caspase-1 expression in GALT and peripheral blood. PLoS ONE 2018, 13, e0192845. [Google Scholar] [CrossRef]
- Alboni, S.; Cervia, D.; Sugama, S.; Conti, B. Interleukin 18 in the CNS. J. Neuroinflamm. 2010, 7, 9. [Google Scholar] [CrossRef]
- Sreeram, S.; Ye, F.; Garcia-Mesa, Y.; Nguyen, K.; El Sayed, A.; Leskov, K.; Karn, J. The potential role of HIV-1 latency in promoting neuroinflammation and HIV-1-associated neurocognitive disorder. Trends Immunol. 2022, 43, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Puricelli, C.; Gigliotti, C.L.; Stoppa, I.; Sacchetti, S.; Pantham, D.; Scomparin, A.; Rolla, R.; Pizzimenti, S.; Dianzani, U.; Boggio, E.; et al. Use of Poly Lactic-co-glycolic Acid Nano and Micro Particles in the Delivery of Drugs Modulating Different Phases of Inflammation. Pharmaceutics 2023, 15, 1772. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Frey, W.H., 2nd. Strategies for intranasal delivery of therapeutics for the prevention and treatment of neuroAIDS. J. Neuroimmune Pharmacol. 2007, 2, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, X.; Meyers, K.; Liu, L.; Su, B.; Wang, P.; Li, Z.; Li, L.; Zhang, T.; Li, N.; et al. Cytokine cascade and networks among MSM HIV seroconverters: Implications for early immunotherapy. Sci. Rep. 2016, 6, 36234. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhou, L.; Zhi, K.; Raji, B.; Pernell, S.; Tadrous, E.; Kodidela, S.; Nookala, A.; Kochat, H.; Kumar, S. Challenges in Biomaterial-Based Drug Delivery Approach for the Treatment of Neurodegenerative Diseases: Opportunities for Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 138. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Godse, S.; Sinha, N.; Kodidela, S.; Singh, U.; Kumar, S. Darunavir Nanoformulation Suppresses HIV Pathogenesis in Macrophages and Improves Drug Delivery to the Brain in Mice. Pharmaceutics 2024, 16, 555. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics16040555
Zhou L, Godse S, Sinha N, Kodidela S, Singh U, Kumar S. Darunavir Nanoformulation Suppresses HIV Pathogenesis in Macrophages and Improves Drug Delivery to the Brain in Mice. Pharmaceutics. 2024; 16(4):555. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics16040555
Chicago/Turabian StyleZhou, Lina, Sandip Godse, Namita Sinha, Sunitha Kodidela, Udai Singh, and Santosh Kumar. 2024. "Darunavir Nanoformulation Suppresses HIV Pathogenesis in Macrophages and Improves Drug Delivery to the Brain in Mice" Pharmaceutics 16, no. 4: 555. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics16040555