
Partial Atomic Model of the Tailed Lactococcal Phage TP901-1 as Predicted by AlphaFold2: Revelations and Limitations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
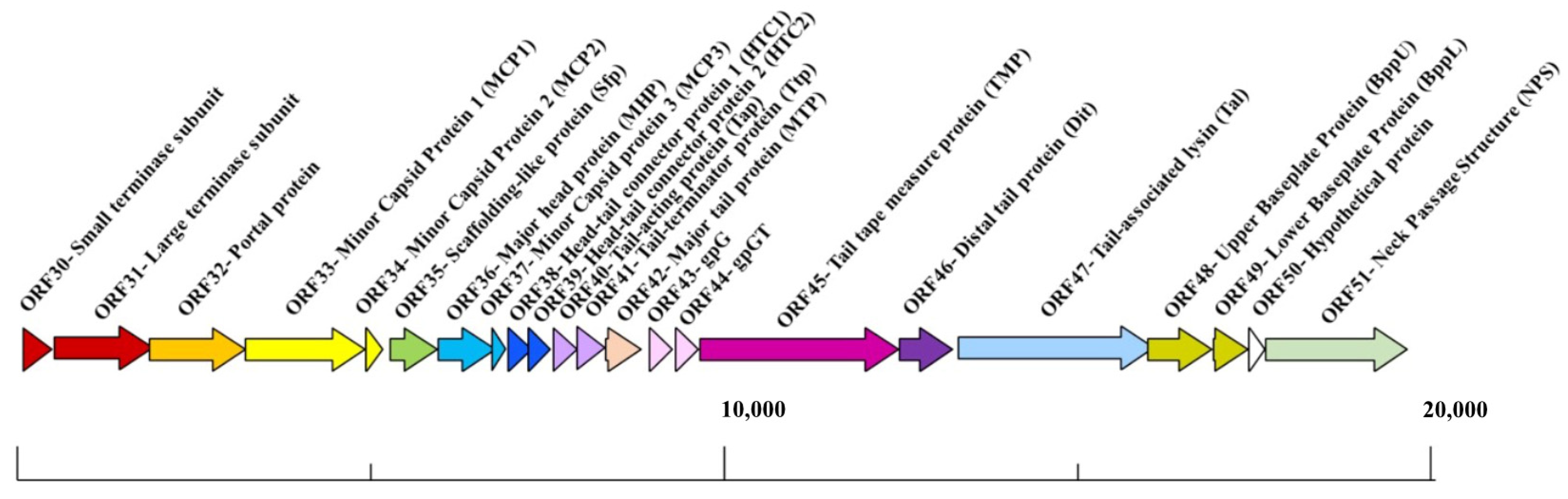
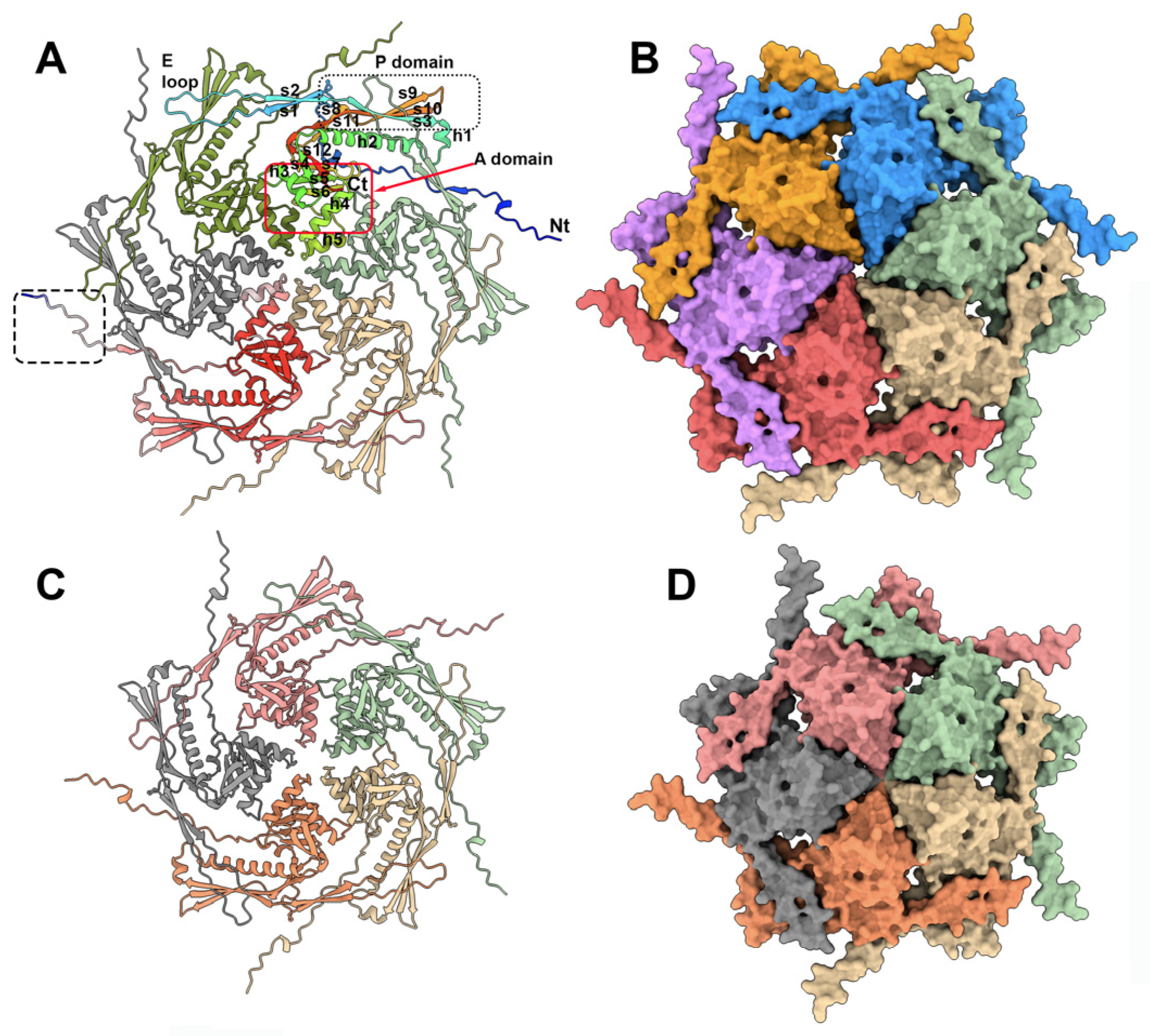
3.1. The TP901-1 Capsid
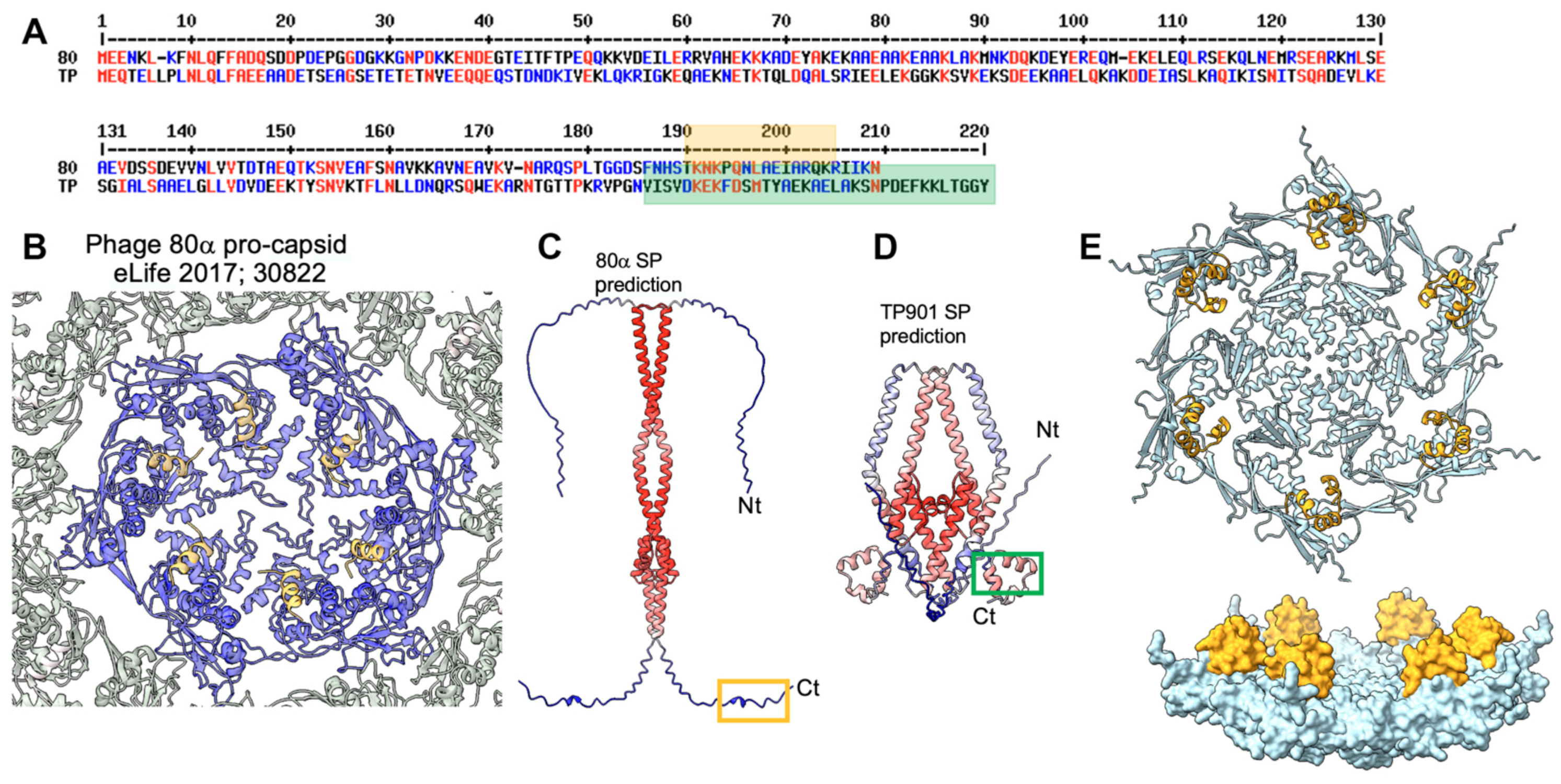
3.2. The Procapsid Scaffolding Protein
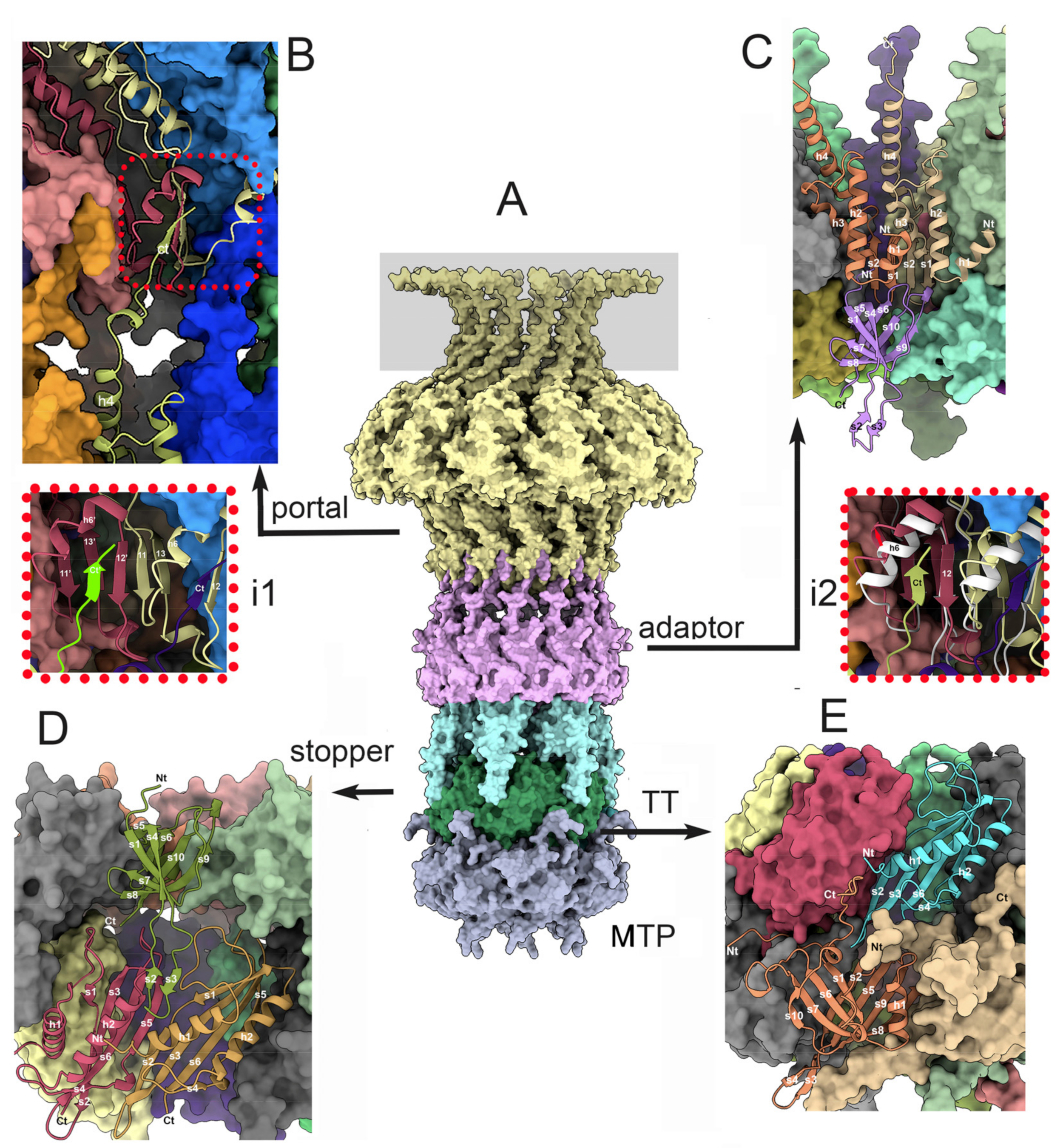
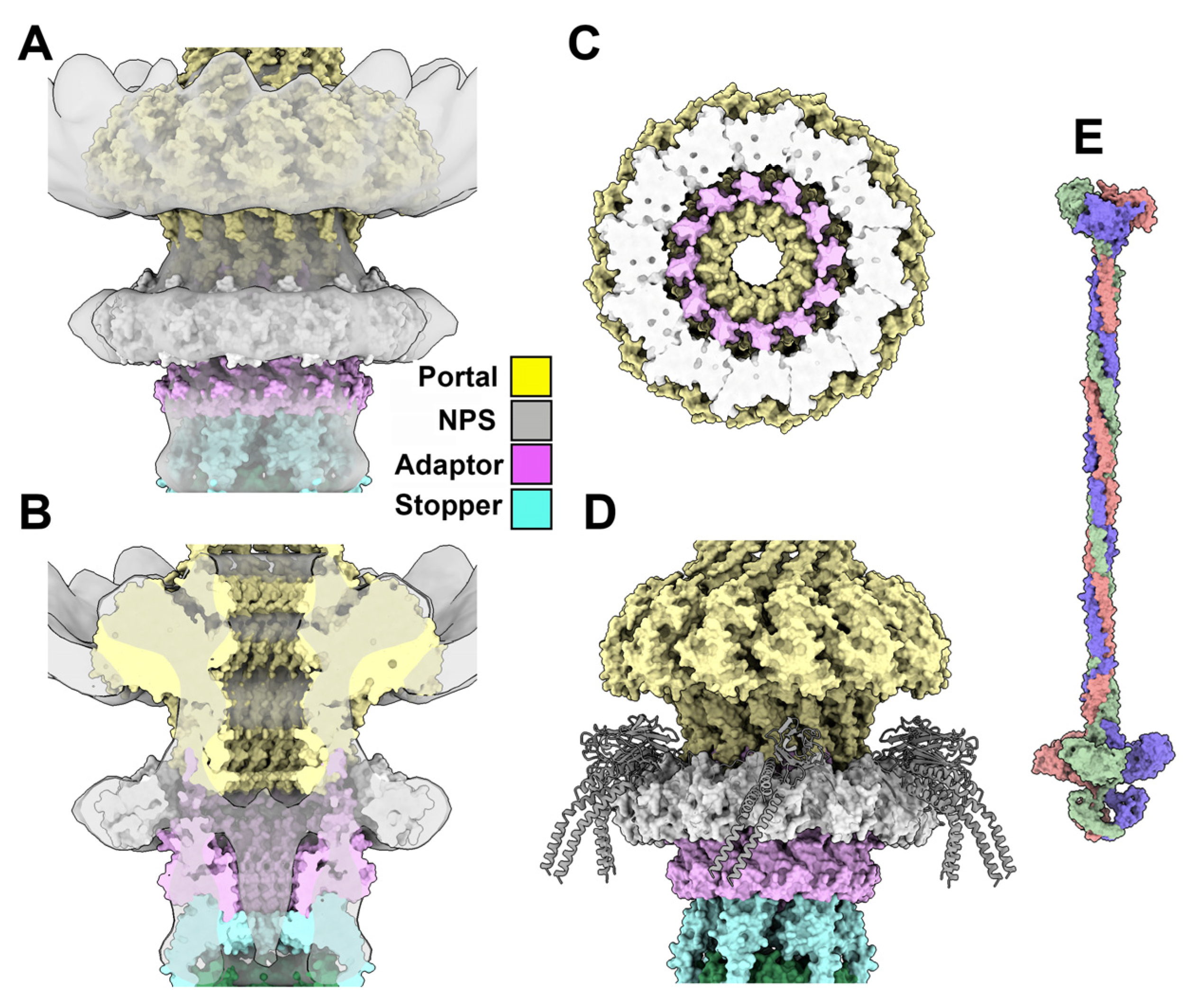
3.3. Dodecameric Portal and Adaptor and the Hexameric Stopper
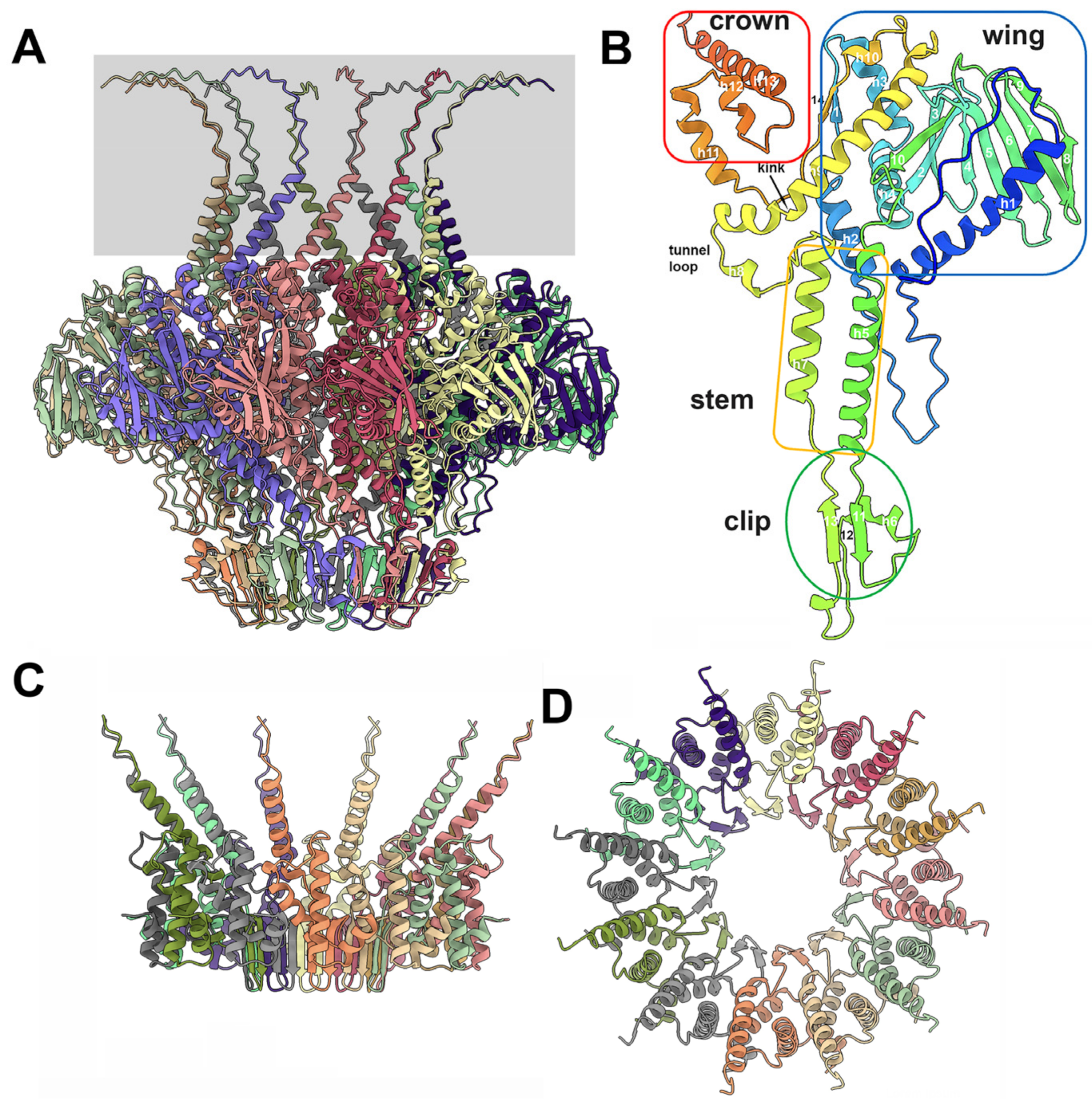
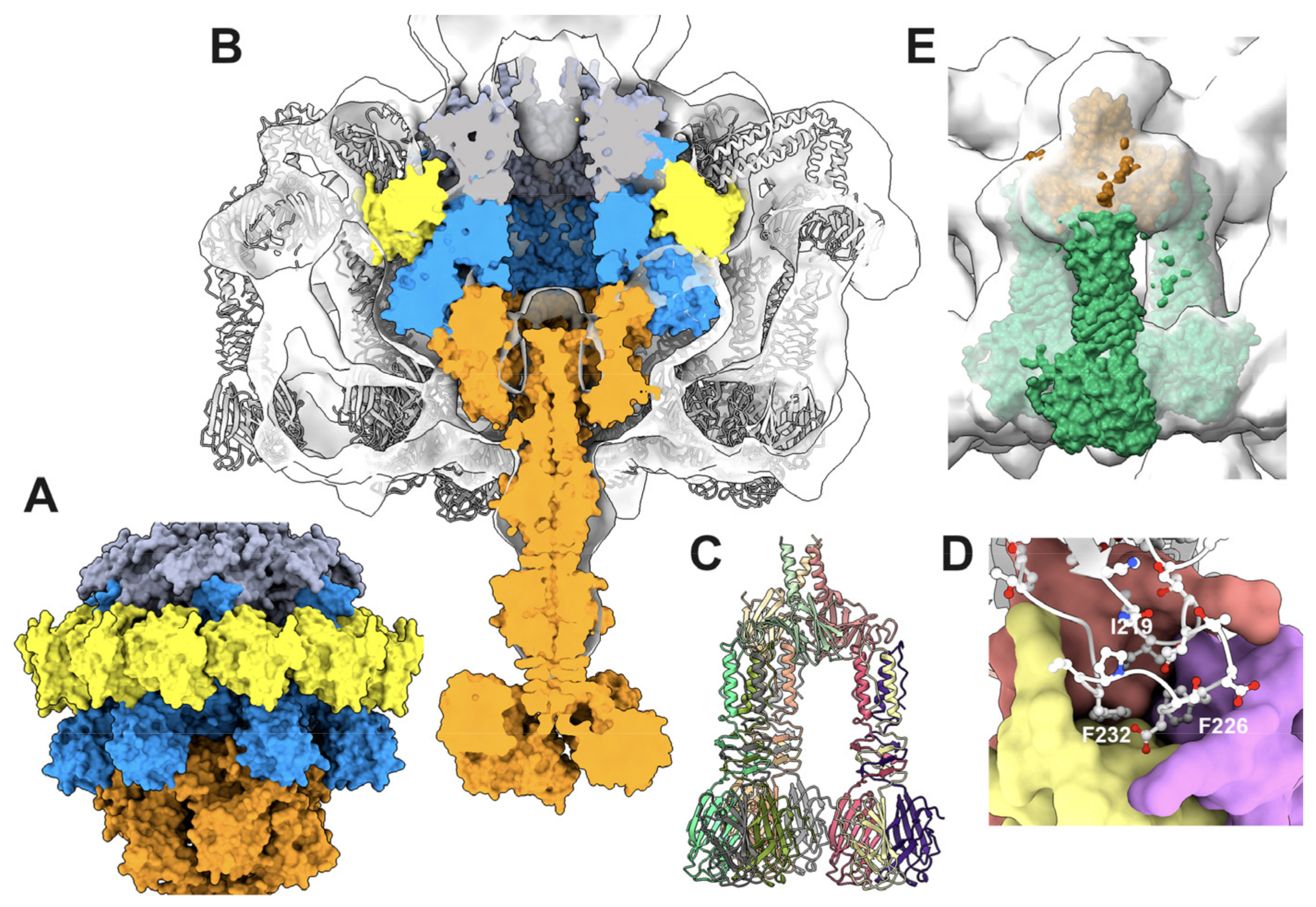
3.3.1. The Dodecameric Portal (ORF32)
3.3.2. The Dodecameric Adaptor (ORF38)
3.3.3. The Portal/Adaptor Complex
3.3.4. The Adaptor/Stopper (ORF39) Complex
3.3.5. The Collar and Neck Passage Proteins
3.4. The Neck/Tail Junction and the Tail
3.4.1. The Stopper/Tail Terminator (TT; ORF41) Complex
3.4.2. The Tail Terminator (TT)/Major Tail Protein (MTP; ORF42) Complex
3.4.3. The MTP/MTP Rings
3.5. The Baseplate
3.5.1. The MTP/Dit Interface
3.5.2. The Trimeric Tal and the Dit/Tal Interface
3.5.3. The Dit/BppU and BppU/RBP Complexes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ackermann, H.W. Bacteriophage Electron Microscopy. Adv. Virus Res. 2012, 82, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Shkoporov, A.N.; Lood, C.; Millard, A.D.; Dutilh, B.E.; Alfenas-Zerbini, P.; van Zyl, L.J.; Aziz, R.K.; Oksanen, H.M.; Adriaenssens, E.M. Abolishment of Morphology-Based Taxa and Change to Binomial Species Names: 2022 Taxonomy Update of the Ictv Bacterial Viruses Subcommittee. Arch. Virol. 2023, 168, 74. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Tremblay, D.; Moineau, S.; Rattei, T.; Kushkina, A.I.; Tovkach, F.I.; Krisch, H.M.; Ackermann, H.W. Phage Morphology Recapitulates Phylogeny: The Comparative Genomics of a New Group of Myoviruses. PLoS ONE 2012, 7, e40102. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Spinelli, S.; Mahony, J.; Cambillau, C. Conserved and Diverse Traits of Adhesion Devices from Siphoviridae Recognizing Proteinaceous or Saccharidic Receptors. Viruses 2020, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Spinelli, S.; Mahony, J.; Lichiere, J.; Blangy, S.; Bricogne, G.; Legrand, P.; Ortiz-Lombardia, M.; Campanacci, V.; van Sinderen, D.; et al. Structure of the Phage TP901-1 1.8 MDa Baseplate Suggests an Alternative Host Adhesion Mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 8954–8958. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, K.; Goulet, A.; McDonnel, B.; Spinelli, S.; van Sinderen, D.; Mahony, J.; Cambillau, C. Revisiting the Host Adhesion Determinants of Streptococcus Thermophilus Siphophages. Microb. Biotech. 2020, 13, 1765–1779. [Google Scholar] [CrossRef] [PubMed]
- Bebeacua, C.; Tremblay, D.; Farenc, C.; Chapot-Chartier, M.P.; Sadovskaya, I.; van Heel, M.; Veesler, D.; Moineau, S.; Cambillau, C. Structure, Adsorption to Host, and Infection Mechanism of Virulent Lactococcal Phage p2. J. Virol. 2013, 87, 12302–12312. [Google Scholar] [CrossRef]
- Baptista, C.; Santos, M.A.; Sao-Jose, C. Phage SPP1 Reversible Adsorption to Bacillus subtilis cell Wall Teichoic Acids Accelerates Virus Recognition of Membrane Receptor YueB. J. Bacteriol. 2008, 190, 4989–4996. [Google Scholar] [CrossRef]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Chaperone-Protein Interactions That Mediate Assembly of the Bacteriophage Lambda Tail to the Correct Length. J. Mol. Biol. 2014, 426, 1004–1018. [Google Scholar] [CrossRef]
- Degroux, S.; Effantin, G.; Linares, R.; Schoehn, G.; Breyton, C. Deciphering Bacteriophage T5 Host Recognition Mechanism and Infection Trigger. J. Virol. 2023, 97, e0158422. [Google Scholar] [CrossRef]
- Mahony, J.; Stockdale, S.R.; Collins, B.; Spinelli, S.; Douillard, F.P.; Cambillau, C.; van Sinderen, D. Lactococcus lactis phage TP901-1 as a model for Siphoviridae virion assembly. Bacteriophage 2016, 6, e1123795. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, M.G. Molecular Architecture of Tailed Double-Stranded DNA Phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Bebeacua, C.; Orlov, I.; Tremblay, D.; Klaholz, B.P.; Moineau, S.; Cambillau, C. Cryo-Electron Microscopy Structure of Lactococcal Siphophage 1358 virion. J. Virol. 2014, 88, 8900–8910. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Joos, R.; Lavelle, K.; van Sinderen, D.; Mahony, J.; Cambillau, C. A structural Discovery Journey of Streptococcal Phages Adhesion Devices by AlphaFold2. Front. Mol. Biosci. 2022, 9, 960325. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Cambillau, C. A Common Evolutionary Origin for Tailed-Bacteriophage Functional Modules and bacterial Machineries. Microbiol. Mol. Biol. Rev. 2011, 75, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Cruz, S.; Erazo Garzon, A.; Kelleher, P.; Bottacini, F.; Breum, S.O.; Neve, H.; Heller, K.J.; Vogensen, F.K.; Palussiere, S.; Courtin, P.; et al. Host Genetic Requirements for DNA Release of Lactococcal Phage TP901-1. Microb. Biotechnol. 2022, 15, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, S.R.; Collins, B.; Spinelli, S.; Douillard, F.P.; Mahony, J.; Cambillau, C.; van Sinderen, D. Structure and Assembly of TP901-1 Virion Unveiled by Mutagenesis. PLoS ONE 2015, 10, e0131676. [Google Scholar] [CrossRef]
- Ostergaard Breum, S.; Neve, H.; Heller, K.J.; Vogensen, F.K. Temperate Phages TP901-1 and phiLC3, Belonging to the P335 Species, Apparently Use Different Pathways for DNA Injection in Lactococcus lactis subsp. cremoris 3107. FEMS Microbiol. Lett. 2007, 276, 156–164. [Google Scholar] [CrossRef]
- Rasmussen, K.K.; Palencia, A.; Varming, A.K.; El-Wali, H.; Boeri Erba, E.; Blackledge, M.; Hammer, K.; Herrmann, T.; Kilstrup, M.; Lo Leggio, L.; et al. Revealing the Mechanism of Repressor Inactivation during Switching of a Temperate Bacteriophage. Proc. Natl. Acad. Sci. USA 2020, 117, 20576–20585. [Google Scholar] [CrossRef]
- Pedersen, M.; Neergaard, J.T.; Cassias, J.; Rasmussen, K.K.; Lo Leggio, L.; Sneppen, K.; Hammer, K.; Kilstrup, M. Repression of the lysogenic P(R) Promoter in Bacteriophage TP901-1 through Binding of a CI-MOR Complex to a Composite O(M)-O(R) Operator. Sci. Rep. 2020, 10, 8659. [Google Scholar] [CrossRef]
- Varming, A.K.; Rasmussen, K.K.; Zong, Z.; Thulstrup, P.W.; Kilstrup, M.; Lo Leggio, L. Flexible linker modulates the binding affinity of the TP901-1 CI phage repressor to DNA. FEBS J. 2022, 289, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Bebeacua, C.; Lai, L.; Vegge, C.S.; Brondsted, L.; van Heel, M.; Veesler, D.; Cambillau, C. Visualizing a Complete Siphoviridae Member by Single-Particle Electron Microscopy: The Structure of Lactococcal Phage TP901-1. J. Virol. 2013, 87, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Vegge, C.S.; Brondsted, L.; Neve, H.; Mc Grath, S.; van Sinderen, D.; Vogensen, F.K. Structural characterization and assembly of the distal tail structure of the temperate lactococcal bacteriophage TP901-1. J. Bacteriol. 2005, 187, 4187–4197. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, S.; Sadovskaya, I.; Vinogradov, E.; Courtin, P.; Guerardel, Y.; Mahony, J.; Grard, T.; Cambillau, C.; Chapot-Chartier, M.P.; van Sinderen, D. Differences in Lactococcal Cell Wall Polysaccharide Structure Are Major Determining Factors in Bacteriophage Sensitivity. MBio 2014, 5, e00880-14. [Google Scholar] [CrossRef] [PubMed]
- Sciara, G.; Bebeacua, C.; Bron, P.; Tremblay, D.; Ortiz-Lombardia, M.; Lichiere, J.; van Heel, M.; Campanacci, V.; Moineau, S.; Cambillau, C. Structure of Lactococcal Phage p2 Baseplate and Its Mechanism of Activation. Proc. Natl. Acad. Sci. USA 2010, 107, 6852–6857. [Google Scholar] [CrossRef] [PubMed]
- Mahony, J.; Alqarni, M.; Stockdale, S.; Spinelli, S.; Feyereisen, M.; Cambillau, C.; Sinderen, D.V. Functional and Structural Dissection of the Tape Measure Protein of Lactococcal Phage TP901-1. Sci. Rep. 2016, 6, 36667. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Applying and Improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Jumper, J.; Hassabis, D. Protein Structure Predictions to Atomic Accuracy with AlphaFold. Nat. Methods 2022, 19, 11–12. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Holm, L.; Kaariainen, S.; Rosenstrom, P.; Schenkel, A. Searching protein structure databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C.; Choi, K.H.; Koti, J.S.; Chipman, P.R.; Anderson, D.L.; Rossmann, M.G. Conservation of the Capsid Structure in tailed dsDNA Bacteriophages: The Pseudoatomic Structure of phi29. Mol. Cell 2005, 18, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Helgstrand, C.; Wikoff, W.R.; Duda, R.L.; Hendrix, R.W.; Johnson, J.E.; Liljas, L. The Refined Structure of a Protein Catenane: The HK97 Bacteriophage Capsid at 3.44 A Resolution. J. Mol. Biol. 2003, 334, 885–899. [Google Scholar] [CrossRef]
- Rao, V.B.; Fokine, A.; Fang, Q.; Shao, Q. Bacteriophage T4 Head: Structure, Assembly, and Genome Packaging. Viruses 2023, 15, 527. [Google Scholar] [CrossRef]
- Kizziah, J.L.; Manning, K.A.; Dearborn, A.D.; Wall, E.A.; Klenow, L.; Hill, R.L.L.; Spilman, M.S.; Stagg, S.M.; Christie, G.E.; Dokland, T. Cleavage and Structural Transitions during Maturation of Staphylococcus aureus Bacteriophage 80α and SaPI1 Capsids. Viruses 2017, 9, 384. [Google Scholar] [CrossRef]
- Ignatiou, A.; Brasiles, S.; El Sadek Fadel, M.; Burger, J.; Mielke, T.; Topf, M.; Tavares, P.; Orlova, E.V. Structural Transitions during the Scaffolding-Driven Assembly of a Viral Capsid. Nat. Commun. 2019, 10, 4840. [Google Scholar] [CrossRef]
- Lebedev, A.A.; Krause, M.H.; Isidro, A.L.; Vagin, A.A.; Orlova, E.V.; Turner, J.; Dodson, E.J.; Tavares, P.; Antson, A.A. Structural Framework for DNA Translocation via the Viral Portal Protein. EMBO J. 2007, 26, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Fokine, A.; Fang, Q. The Remarkable Viral Portal Vertex: Structure and a Plausible Model for Mechanism. Curr. Opin. Virol. 2021, 51, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Feiss, M.; Rao, V.B. The Bacteriophage DNA Packaging Machine. Adv. Exp. Med. Biol. 2012, 726, 489–509. [Google Scholar] [CrossRef] [PubMed]
- Orlov, I.; Roche, S.; Brasiles, S.; Lukoyanova, N.; Vaney, M.C.; Tavares, P.; Orlova, E.V. CryoEM Structure and Assembly Mechanism of a Bacterial Virus Genome Gatekeeper. Nat. Commun. 2022, 13, 7283. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Fabrega-Ferrer, M.; Machon, C.; Conesa, J.J.; Fernandez, F.J.; Perez-Luque, R.; Perez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 Bacteriophage Portal and Tail Suggest a Viral DNA Retention and Ejection Mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef] [PubMed]
- Bardy, P.; Fuzik, T.; Hrebik, D.; Pantucek, R.; Thomas Beatty, J.; Plevka, P. Structure and Mechanism of DNA Delivery of a Gene Transfer Agent. Nat. Commun. 2020, 11, 3034. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Mahony, J.; Cambillau, C.; van Sinderen, D. Exploring Structural Diversity among Adhesion Devices Encoded by Lactococcal P335 Phages with AlphaFold2. Microorganisms 2022, 10, 2278. [Google Scholar] [CrossRef]
- Kizziah, J.L.; Manning, K.A.; Dearborn, A.D.; Dokland, T. Structure of the Host Cell Recognition and Penetration Machinery of a Staphylococcus aureus Bacteriophage. PLoS Pathog. 2020, 16, e1008314. [Google Scholar] [CrossRef]
- Zinke, M.; Sachowsky, K.A.A.; Oster, C.; Zinn-Justin, S.; Ravelli, R.; Schroder, G.F.; Habeck, M.; Lange, A. Architecture of the Flexible Tail Tube of Bacteriophage SPP1. Nat. Commun. 2020, 11, 5759. [Google Scholar] [CrossRef]
- Labrie, S.J.; Josephsen, J.; Neve, H.; Vogensen, F.K.; Moineau, S. Morphology, Genome Sequence, and Structural Proteome of type Phage P335 from Lactococcus lactis. Appl. Environ. Microbiol. 2008, 74, 4636–4644. [Google Scholar] [CrossRef]
- Mahony, J.; Oliveira, J.; Collins, B.; Haanemaaijer, L.; Lugli, G.A.; Neve, H.; Ventura, M.; Kouwen, T.R.; Cambillau, C.; van Sinderen, D.; et al. Genetic and Functional Characterisation of the Lactococcal P335 Phage-Host Interactions. BMC Genom. 2017, 18, 146. [Google Scholar] [CrossRef]
- Veesler, D.; Robin, G.; Lichiere, J.; Auzat, I.; Tavares, P.; Bron, P.; Campanacci, V.; Cambillau, C. Crystal Structure of Bacteriophage SPP1 Distal Tail Protein (gp19.1): A Baseplate Hub Paradigm in Gram-Positive Infecting Phages. J. Biol. Chem. 2010, 285, 36666–36673. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, S.; Leiman, P.G.; Kostyuchenko, V.A.; Chipman, P.R.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure of the Cell-Puncturing Device of Bacteriophage T4. Nature 2002, 415, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 Baseplate and Its Function in Triggering Sheath Contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef]
- Stockdale, S.R.; Mahony, J.; Courtin, P.; Chapot-Chartier, M.P.; van Pijkeren, J.P.; Britton, R.A.; Neve, H.; Heller, K.J.; Aideh, B.; Vogensen, F.K.; et al. The lactococcal phages Tuc2009 and TP901-1 Incorporate Two Alternate Forms of Their Tail Fiber into Their Virions for Infection Specialization. J. Biol. Chem. 2013, 288, 5581–5590. [Google Scholar] [CrossRef] [PubMed]
- Linares, R.; Arnaud, C.A.; Effantin, G.; Darnault, C.; Epalle, N.H.; Boeri Erba, E.; Schoehn, G.; Breyton, C. Structural Basis of Bacteriophage T5 Infection Trigger and E. coli Cell Wall Perforation. Sci. Adv. 2023, 9, eade9674. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C.; Kanamaru, S.; Badasso, M.O.; Koti, J.S.; Owen, B.A.; McMurray, C.T.; Anderson, D.L.; Rossmann, M.G. BACTERIOPHAGE phi29 Scaffolding Protein gp7 before and after Prohead Assembly. Nat. Struct. Biol. 2003, 10, 572–576. [Google Scholar] [CrossRef]
- Li, S.; Roy, P.; Travesset, A.; Zandi, R. Why Large Icosahedral Viruses Need Scaffolding Proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 10971–10976. [Google Scholar] [CrossRef]
- Dearborn, A.D.; Wall, E.A.; Kizziah, J.L.; Klenow, L.; Parker, L.K.; Manning, K.A.; Spilman, M.S.; Spear, J.M.; Christie, G.E.; Dokland, T. Competing Scaffolding Proteins Determine Capsid Size during Mobilization of Staphylococcus aureus Pathogenicity Islands. eLife 2017, 6, e30822. [Google Scholar] [CrossRef]
- Orlova, E.V.; Dube, P.; Beckmann, E.; Zemlin, F.; Lurz, R.; Trautner, T.A.; Tavares, P.; van Heel, M. Structure of the 13-Fold Symmetric Portal Protein of Bacteriophage SPP1. Nat. Struct. Biol. 1999, 6, 842–846. [Google Scholar]
- Cuervo, A.; Vaney, M.C.; Antson, A.A.; Tavares, P.; Oliveira, L. Structural Rearrangements between Portal Protein Subunits Are Essential for Viral DNA Translocation. J. Biol. Chem. 2007, 282, 18907–18913. [Google Scholar] [CrossRef]
- Linares, R.; Arnaud, C.A.; Degroux, S.; Schoehn, G.; Breyton, C. Structure, Function and Assembly of the Long, Flexible Tail of Siphophages. Curr. Opin. Virol. 2020, 45, 34–42. [Google Scholar] [CrossRef]
- Arnaud, C.A.; Effantin, G.; Vives, C.; Engilberge, S.; Bacia, M.; Boulanger, P.; Girard, E.; Schoehn, G.; Breyton, C. Bacteriophage T5 Tail Tube Structure Suggests a Trigger Mechanism for Siphoviridae DNA ejection. Nat. Commun. 2017, 8, 1953. [Google Scholar] [CrossRef]
- Cambillau, C.; Goulet, A. Exploring Host-Binding Machineries of Mycobacteriophages with AlphaFold2. J. Virol. 2023, 97, e0179322. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahony, J.; Goulet, A.; van Sinderen, D.; Cambillau, C. Partial Atomic Model of the Tailed Lactococcal Phage TP901-1 as Predicted by AlphaFold2: Revelations and Limitations. Viruses 2023, 15, 2440. https://0-doi-org.brum.beds.ac.uk/10.3390/v15122440
Mahony J, Goulet A, van Sinderen D, Cambillau C. Partial Atomic Model of the Tailed Lactococcal Phage TP901-1 as Predicted by AlphaFold2: Revelations and Limitations. Viruses. 2023; 15(12):2440. https://0-doi-org.brum.beds.ac.uk/10.3390/v15122440
Chicago/Turabian StyleMahony, Jennifer, Adeline Goulet, Douwe van Sinderen, and Christian Cambillau. 2023. "Partial Atomic Model of the Tailed Lactococcal Phage TP901-1 as Predicted by AlphaFold2: Revelations and Limitations" Viruses 15, no. 12: 2440. https://0-doi-org.brum.beds.ac.uk/10.3390/v15122440