3.1.2. Synthesis of Hybrids 6a–l
A mixture of carbohydrazide 4 (0.196 g, 1.0 mmol) and indoline-2,3-dione derivatives 5a–l (1.0 mmol) in EtOH (30 mL) and AcOH (0.3 mL) was refluxed for 4 h. Crystallization of the resulting solid from EtOH/DMF gave compounds 6a–l, respectively.
6-Methyl-N′-(2-oxoindolin-3-ylidene)imidazo[2,1-b]thiazole-5-carbohydrazide (6a)
Yield = 68%; m.p. = >300 °C; IR: 1697 (>C=O), 3263 (>N-H), 3444 (>N-H); 1H NMR: 2.65 (s, 3H, -CH3), 6.92 (d, J = 8.1 Hz, 1H, Ar-H), 7.07 (d, J = 8.1 Hz, 1H, Ar-H), 7.31–7.36 (m, 1H, Ar-H), 7.41 (d, J = 4.5 Hz, 1H, Ar-H), 7.53 (d, J = 7.5 Hz, 1H, Ar-H), 8.21 (d, J = 4.5 Hz, 1H, Ar-H), 11.11 (s, 1H, >N-H), 13.26 (s, 1H, >N-H); 13C NMR: 16.88 (-CH3), 111.73, 115.41, 117.66, 120.61, 121.22, 121.74, 132.04, 137.52, 142.82, 148.80, 152.63, 157.08 (>C=O), 163.16 (>C=O); Anal. for C15H11N5O2S (325.35): calcd.: C, 55.38; H, 3.41; N, 21.53; found: C, 55.50; H, 3.47; N, 21.37.
N′-(5-Fluoro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6b)
Yield = 74%; m.p. = >300 °C; IR: 1710 (>C=O), 3294 (>N-H), 3420 (>N-H); 1H NMR: 2.65 (s, 3H, -CH3), 6.93 (q, J = 4.0 Hz, 1H, Ar-H), 7.15–7.19 (m, 1H, Ar-H), 7.33 (dd, J = 8.1 Hz, 1H, Ar-H), 7.42 (d, J = 4.1 Hz, 1H, Ar-H), 8.22 (d, J = 4.0 Hz, 1H, Ar-H), 11.14 (s, 1H, >N-H), 13.28 (s, 1H, >N-H); 13C NMR: 16.89 (-CH3), 40.73, 108.34, 112.85, 115.54 (2C), 117.59, 118.29, 121.75 (2C), 139.05, 149.06, 152.81, 157.01 (>C=O), 163.29 (>C=O); Anal. for C15H10FN5O2S (343.34): calcd.: C, 52.47; H, 2.94; N, 20.40; found: C, 52.21; H, 2.99; N, 20.53.
N′-(5-Chloro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6c)
Yield = 67%; m.p. = 292–293 °C; IR: 1705 (>C=O), 3124 (>N-H), 3367 (>N-H); 1H NMR: 2.68 (s, 3H, -CH3), 6.95 (d, J = 8.5 Hz, 1H, Ar-H), 7.39 (d, J = 4.0 Hz, 1H, Ar-H), 7.48 (d, J = 4.0 Hz, 1H, Ar-H), 7.51 (m, 1H, Ar-H), 8.26 (d, J = 4.4 Hz, 1H, Ar-H),11.38 (s, 1H, >N-H), 13.26 (s, 1H, >N-H); 13C NMR: 16.91 (-CH3), 113.25, 115.53, 117.56, 120.67, 121.75, 122.31, 127.42, 131.39, 136.48, 141.44, 149.03, 152.80, 156.92 (>C=O), 162.94 (>C=O); Anal. for C15H10ClN5O2S (359.79): calcd.: C, 50.08; H, 2.80; N, 19.47; found: C, 49.91; H, 3.01; N, 19.50.
N′-(5-Bromo-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6d)
Yield = 66%; m.p. = 296–297 °C; IR: 1705 (>C=O), 3116 (>N-H), 3383 (>N-H); 1H NMR: 2.62 (s, 3H, -CH3), 6.85 (s, 1H, Ar-H), 7.42–7.46 (m, 2H, Ar-Hs), 7.56 (s, 1H, Ar-H), 8.20 (s, 1H, Ar-H), 11.33 (s, 1H, >N-H), 13.20 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.07 (2C), 115.59, 115.76 (2C), 118.58, 118.98, 1120.82, 136.62, 137.29, 144.84, 154.29, 156.98 (C=O), 158.87 (C=O); Anal. for C15H10BrN5O2S (404.24): calcd.: C, 44.57; H, 2.49; N, 17.33; found: C, 44.30; H, 2.36; N, 17.37.
6-Methyl-N′-(2-oxo-1-propylindolin-3-ylidene)imidazo[2,1-b]thiazole-5-carbohydrazide (6e)
Yield = 51%; m.p. = 256–257 °C; IR: 1702 (>C=O), 3236 (>N-H); 1H NMR: 0.89 (t, J = 7.5 Hz, 3H, -CH3), 1.62–1.69 (m, 2H, >CH2), 2.67 (s, 3H, -CH3), 3.71 (t, J = 7.5 Hz, 2H, >CH2), 7.12–7.19 (m, 2H, Ar-Hs), 7.41–7.44 (m, 2H, Ar-Hs), 7.59 (d, J = 8.1 Hz, 1H, Ar-H), 8.22 (d, J = 4.0 Hz, 1H, Ar-H), 13.21 (s, 1H, >N-H); 13C NMR: 11.55 (-CH3), 17.03 (-CH3), 28.89 (>CH2), 41.65 (>CH2), 112.80, 113.01, 115.65, 121.82, 123.48, 134.33, 135.65, 142.20, 148.27, 152.71, 156.25, 161.10 (>C=O); Anal. for C18H17N5O2S (367.43): calcd.: C, 58.84; H, 4.66; N, 19.06; found: C, 59.07; H, 4.59; N, 19.20.
N′-(5-Fluoro-2-oxo-1-propylindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6f)
Yield = 66%; m.p. = 243–244 °C; IR: 1689 (>C=O), 3224 (>N-H); 1H NMR: 0.92 (t, J = 7.5 Hz, 3H, -CH3), 1.64–1.70 (m, 2H, >CH2), 2.72 (s, 3H, -CH3), 3.76 (t, J = 7.5 Hz, 2H, >CH2), 7.26–7.32 (m, 2H, Ar-Hs), 7.43–7.45 (m, 1H, Ar-H), 7.48 (d, J = 4.4 Hz, 1H, Ar-H), 8.38 (s, 1H, Ar-H), 13.21 (s, 1H, >N-H); 13C NMR: 11.51 (-CH3), 17.20 (-CH3), 29.90 (>CH2), 41.71 (>CH2), 113.21, 113.29, 114.50, 123.18, 125.58, 132.10, 134.23, 143.60, 149.55, 153.79, 156.63, 162.12 (>C=O); Anal. for C18H16FN5O2S (385.42): calcd.: C, 56.09; H, 4.18; N; 18.17; found: C, 55.82; H, 4.21; N; 18.30.
N′-(5-Chloro-2-oxo-1-propylindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6g)
Yield = 61%; m.p. = 254–255 °C; IR: 1693 (>C=O), 3236 (>N-H); 1H NMR: E/Z, 0.83, 0.87 (2t, J = 7.0 Hz, J = 7.5 Hz, 3H, -CH3), 1.54–1.63 (2m, 2H, >CH2), 2.52, 2.68 (2s, 3H, -CH3), 3.66, 3.72 (2t, J = 7.0 Hz, J = 7.5 Hz, 2H, >CH2), 7.18, 7.26 (2d, J = 8.5 Hz, J = 9.0 Hz, 1H, Ar-H), 7.39–7.45 (2d, J = 4.0 Hz, J = 4.4 Hz, 1H, Ar-H), 7.48–7.55 (m, 2H, Ar-Hs), 8.18, 8.25 (2s, 1H, Ar-H), 11.46, 13.20 (2s, 1H, >N-H); 13C NMR: 11.43 (-CH3), 16.30 (-CH3), 28.40 (>CH2), 43.51 (>CH2), 112.56, 113.65, 118.58, 124.29, 124.52, 132.18, 134.23, 144.64, 149.05, 154.59, 155.83, 162.23(>C=O); Anal. for C18H16ClN5O2S (401.87): calcd.: C, 53.80; H, 4.01; N, 17.43; found: C, 53.53; H, 3.84; N, 17.57.
N′-(5-Bromo-2-oxo-1-propylindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6h)
Yield = 50%; m.p. = 261–262 °C; IR: 1724 (>C=O), 3232 (>N-H); 1H NMR: E/Z, 0.83, 0.86 (2t, J = 7.0 Hz, J = 7.5 Hz, 3H, -CH3), 1.54–1.63 (2m, 2H, >CH2), 2.36, 2.66 (2s, 3H, -CH3), 3.65, 3.69 (2t, J = 7.0 Hz, J = 7.5 Hz, 2H, >CH2), 7.12, 7.18 (2d, J = 8.5 Hz, J = 9.0 Hz, 1H, Ar-H), 7.38, 7.44 (2d, J = 4.0 Hz, J = 4.5 Hz, 1H, Ar-H), 7.57–7.64 (m, 2H, Ar-Hs), 8.06, 8.23 (2d, J = 4.0 Hz, J = 5.0 Hz, 1H, ArH), 11.14, 13.60 (2s, D2O exchangeable, 1H, >N-H); 13C NMR: 11.58 (-CH3), 17.00 (-CH3), 28.87 (>CH2), 41.60 (>CH2), 112.75, 115.47, 115.60, 121.78, 123.28, 134.10, 135.43, 142.50, 149.17, 152.89, 156.92, 161.04 (>C=O); Anal. for C18H16BrN5O2S (446.32): calcd.: C, 48.44; H, 3.61; N, 15.69; found: C, 48.56; H, 3.53; N, 15.71.
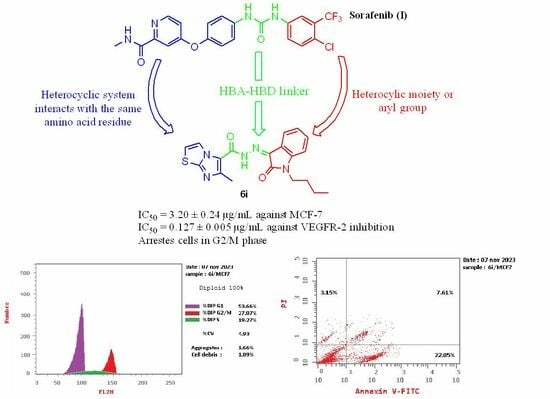
N′-(1-Butyl-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6i)
Yield = 45%; m.p. = 250–251 °C; IR: 1670 (>C=O), 3240 (>N-H); 1H NMR: 0.92 (t, J = 7.2 Hz, 3H, -CH3), 1.35–1.39 (m, 2H, >CH2), 1.60–1.66 (m, 2H, >CH2), 2.73 (s, 3H, -CH3), 3.79 (t, J = 7.3 Hz, 2H, >CH2), 7.17–7.24 (m, 2H, Ar-Hs), 7.45–7.49 (m, 2H, Ar-Hs), 7.64 (d, J= 7.2 Hz, 1H, Ar-H), 8.34 (s, 1H, Ar-H): 13C NMR: 14.02 (-CH3), 16.97 (-CH3), 20.04 (>CH2), 29.64 (>CH2), 39.24 (>CH2), 4077, 42.23, 110.65, 115.48, 117.63, 120.02, 121.06, 121.76, 123.66, 132.02, 136.63, 143.38, 148.91, 161.32 (>C=O); Anal. for C19H19N5O2S (381.45): calcd.: C, 59.83; H, 5.02; N, 18.36; found: C, 59.67; H, 4.85; N, 18.39.
N′-(1-Butyl-5-fluoro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6j)
Yield = 50%; m.p. = 237–238 °C; IR: 1670 (>C=O), 3244 (>N-H); 1H NMR: 0.86 (t, J = 7.0 Hz, 3H, -CH3), 1.26–1.33 (m, 2H, >CH2), 1.53–1.59 (m, 2H, >CH2), 2.66 (s, 3H, -CH3), 3.71 (t, J = 7.5 Hz, 2H, >CH2), 7.17–7.20 (m, 1H, Ar-H), 7.23–7.27 (m, 1H, Ar-H), 7.37 (d, J = 8.1 Hz, 1H, Ar-H), 7.44 (d, J = 4.0 Hz, 1H, Ar-H), 8.22 (d, J = 4.0 Hz, 1H, Ar-H), 13.20 (s, 1H, >N-H); 13C NMR: 14.00 (-CH3), 16.97 (-CH3), 20.01 (>CH2), 29.56 (>CH2), 39.74 (>CH2), 108.16, 108.36, 111.94, 115.59, 117.56, 118.07, 121.43, 136.13, 139.60, 149.14, 152.88, 156.97, 158.35, 160.26, 161.35 (>C=O); Anal. for C19H18FN5O2S (399.44): calcd.: C, 57.13; H, 4.54; N, 17.53; found: C, 57.26; H, 4.57; N, 17.80.
N′-(1-Butyl-5-chloro-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6k)
Yield = 67%; m.p. = 224–225 °C; IR: 1693 (>C=O), 3224 (>N-H); 1H NMR: 0.85 (t, J = 7.5 Hz, 3H, -CH3), 1.27–1.32 (m, 2H, >CH2), 1.55–1.58 (m, 2H, >CH2), 2.68 (s, 3H, -CH3), 3.74 (t, J = 7.5 Hz, 2H, >CH2), 7.24 (d, J = 8.1 Hz, 1H, Ar-H), 7.46–7.49 (m, 2H, Ar-Hs), 7.56 (s, 1H, Ar-H), 8.25 (d, J = 4.0 Hz, 1H, Ar-H), 13.19 (s, 1H, >N-H); 13C NMR: 14.02 (-CH3), 15.99 (-CH3), 20.00 (>CH2), 30.56 (>CH2), 40.52 (>CH2), 43.69, 44.96, 118.70, 121.10, 122.18, 125.52, 126.31, 127.96, 130.24, 138.18, 144.56, 149.16, 157.09, 161.58 (>C=O); Anal. for C19H18ClN5O2S (415.90): calcd.: C, 54.87; H, 4.36; N, 16.84; found: C, 55.06; H, 4.38; N, 17.00.
N′-(5-Romo-1-butyl-2-oxoindolin-3-ylidene)-6-methylimidazo[2,1-b]thiazole-5-carbohydrazide (6l)
Yield = 39%; m.p. = 237–238 °C; IR: 1693 (>C=O), 3228 (>N-H); 1H NMR: 0.86 (t, J = 7.1 Hz, 3H, -CH3), 1.27–1.33 (m, 2H, >CH2), 1.53–1.57 (m, 2H, >CH2), 2.66 (s, 3H, -CH3), 3.71 (t, J = 7.5 Hz, 2H, >CH2), 7.16 (s, 1H, Ar-H), 7.44 (d, J = 5.0 Hz, 1H, Ar-H), 7.58 (s, J = 9.5 Hz, 1H, Ar-H), 7.63 (s, 1H, Ar-H), 8.22 (s, 1H, Ar-H), 13.14 (s, 1H, >N-H); 13C NMR: 14.00 (-CH3), 16.99 (-CH3), 20.00 (>CH2), 29.56 (>CH2), 40.52 (>CH2), 40.69, 40.76, 112.72, 115.48, 115.61, 117.55, 121.78, 123.30, 134.13, 142.42, 149.18, 152.90, 156.93, 160.97 (>C=O); Anal. for C19H18BrN5O2S (460.35): calcd.: C, 49.57; H, 3.94; N, 15.21; found: C, 49.52; H, 4.08; N, 15.34.
3.1.3. Synthesis of Urea Derivatives 8a–g
Compound
4 was stirred for 3 h in a solution of NaNO
2 with HCl to form the 6-methylimidazo[2,1-
b]thiazole-5-carbonyl azide [
41]. Azide
7 (0.21 g, 1 mmol) was refluxed in 1,4-dioxane (40 mL) with different anilines (1.0 mmol) for 8 h. Crystallization from EtOH for the formed solid gave the targeted urea derivatives
8a–
g, respectively.
1-(6-Methylimidazo[2,1-b]thiazol-5-yl)-3-phenylurea (8a)
Yield = 54%; m.p. = 287–288 °C; IR: 1639 (>C=O), 3263 (>N-H), 3290 (>N-H); 1H NMR: 2.09 (s, 3H, -CH3), 6.92 (t, J = 7.5 Hz, 1H, Ar-H), 7.08 (d, J = 4.5 Hz, 1H, Ar-H), 7.22 (t, J = 7.4 Hz, 1H, Ar-H), 7.42 (d, J = 7.8 Hz, 2H, Ar-Hs), 7.53 (d, J = 5.0 Hz, 1H, Ar-H), 8.09 (s, 1H, NH), 8.93 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.05, 118.63, 118.98 (3C), 122.48, 129.21 (2C), 137.23, 140.25, 144.81, 154.18 (>C=O); Anal. for C13H12N4OS (272.33): calcd.: C, 57.34; H, 4.44; N, 20.57; found: C, 57.17; H, 4.26; N, 20.79.
1-(6-Methylimidazo[2,1-b]thiazol-5-yl)-3-(p-tolyl)urea (8b)
Yield = 47%; m.p. = 282–283 °C; IR: 1701 (>C=O), 3267 (>N-H), 3360 (>N-H); 1H NMR: 2.14 (s, 3H, -CH3), 2.25 (s, 3H, -CH3), 7.08 (d, J = 8.0 Hz, 2H, Ar-Hs), 7.13 (d, J = 4.4 Hz, 1H, Ar-H), 7.36 (d, J = 7.8 Hz, 2H, Ar-Hs), 7.57 (d, J = 4.4 Hz, 1H, Ar-H), 8.09 (s, 1H, >N-H), 8.83 (s, 1H, >N-H); 13C NMR: 13.35 (-CH3), 20.87 (-CH3), 112.05, 118.70, 118.98, 119.13 (2C), 129.60 (2C), 131.32, 137.19, 137.65, 144.79, 154.20 (>C=O); Anal. for C14H14N4OS (286.35): calcd.: C, 58.72; H, 4.93; N, 19.57; found: C, 58.66; H, 5.10; N, 19.59.
1-(4-Methoxyphenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8c)
Yield = 50%; m.p. = 284–285 °C; IR: 1639 (>C=O), 3140 (>N-H), 3278 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 3.66 (s, 3H, -O-CH3), 6.80 (d, J = 8.5 Hz, 2H, ArH), 7.08 (d, J = 4.5 Hz, 1H, Ar-H), 7.32 (d, J = 8.5 Hz, 2H, Ar-Hs), 7.52 (d, J = 5.0 Hz, 1H, Ar-H), 8.04 (s, 1H, >N-H), 8.74 (s, 1H, >N-H); 13C NMR: 13.37 (-CH3), 55.67 (-O-CH3), 111.98, 114.37 (2C), 118.83, 120.6 (2C), 133.29, 137.13 144.73, 144.73, 154.37, 155.05 (>C=O); Anal. for C14H14N4O2S (302.35): calcd.: C, 55.62; H, 4.67; N, 18.53; found: C, 55.35; H, 4.69; N, 18.45.
1-(4-Fluorophenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8d)
Yield = 56%; m.p. = 281–282 °C; IR (KBr): 1635 (>C=O), 3163 (>N-H), 3278 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 7.04–7.10 (m, 3H, Ar-Hs), 7.42–7.44 (m, 2H, Ar-Hs), 7.54 (d, J = 5.0 Hz, 1H, Ar-H), 8.12 (s, 1H, >N’-H), 8.98 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.07, 115.59 (2C), 115.76 (2C), 118.58, 118.98, 120.82, 136.62, 137.29, 144.84, 154.29 (>C=O); Anal. for C13H11FN4OS (290.32): calcd.: C, 53.78; H, 3.82; N, 19.30; found: C, 53.80; H, 4.02; N, 19.53.
1-(4-Chlorophenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8e)
Yield = 48%; m.p. = 285–286 °C; IR: 1708 (>C=O), 3267 (>N-H), 3360 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 7.09 (d, J = 4.0 Hz, 1H, Ar-H), 7.26 (d, J = 9.0 Hz, 2H, Ar-Hs), 7.45 (d, J = 8.5 Hz, 1H, Ar-H), 7.54 (d, J = 4.5 Hz, 1H, Ar-H), 8.19 (s, 1H, >N-H), 9.12 (s, 1H, >N-H); 13C NMR: 13.36 (-CH3), 112.10, 118.46, 118.99, 120.56 (2C), 126.00, 129.03 (2C), 137.35, 139.32, 144.89, 154.13 (>C=O); Anal. for C13H11ClN4OS (306.77): calcd.: C, 50.90; H, 3.61; N, 18.26; found: C, 50.73; H, 3.84; N, 18.39.
1-(4-Bromophenyl)-3-(6-methylimidazo[2,1-b]thiazol-5-yl)urea (8f)
Yield = 50%; m.p. = 288–289 °C; IR: 1709 (>C=O), 3263 (>N-H), 3360 (>N-H); 1H NMR: 2.08 (s, 3H, -CH3), 7.09 (d, J = 4.5 Hz, 1H, Ar-H), 7.37–7.42 (m, 4H, Ar-Hs), 7.55 (d, J = 4.5 Hz, 1H, Ar-H), 8.27 (s, 1H, >N-H), 9.20 (s, 1H, >N-H); 13C NMR: 13.37 (-CH3), 112.09, 113.87, 118.48, 119.00, 120.96 (2C), 131.92 (2C), 137.30, 139.79, 144.87, 154.12 (>C=O); Anal. for:C13H11BrN4OS (351.22): calcd.: C, 44.46; H, 3.16; N, 15.95; found: C, 44.68; H, 3.28; N, 16.17.
4-(3-(6-Methylimidazo[2,1-b]thiazol-5-yl)ureido)benzenesulfonamide (8g)
Yield = 56%; m.p. = 283–284 °C; IR: 1708 (>C=O), 3120 (>N-H), 3275 (>N-H); 1H NMR: 2.09 (s, 3H, -CH3), 7.09 (d, J = 4.5 Hz, 1H, Ar-H), 7.17 (s, 2H, -NH2), 7.58 (d, J = 9.0 Hz, 1H, Ar-H), 7.57 (t, J = 8.5 Hz, 3H, Ar-Hs), 7.68 (d, J = 8.5 Hz, 2H, Ar-Hs), 8.26 (s, 1H, >N-H), 9.35 (s, 1H, >N-H); 13C NMR: 13.37 (-CH3), 112.18, 118.26, 118.32 (2C), 119.01, 127.21 (2C), 137.39, 137.55, 143.42, 144.98, 153.99 (>C=O); Anal. for C13H13N5O3S2 (351.40); calcd.: C, 44.43; H, 3.73; N, 19.93; found: C, 44.25; H, 3.56; N, 20.11.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
