
Synthesis of Mixed Phosphonate Esters and Amino Acid-Based Phosphonamidates, and Their Screening as Herbicides
 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
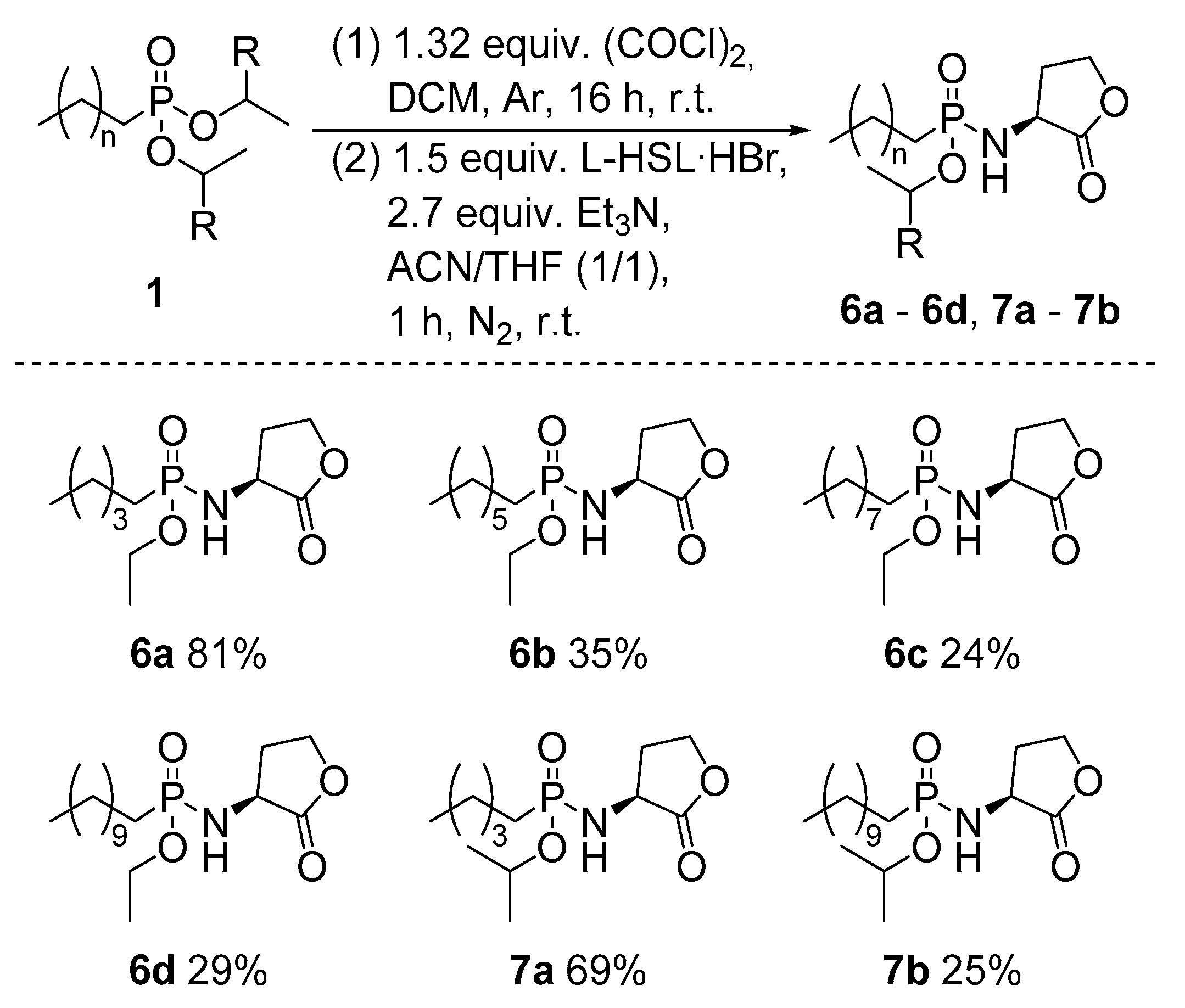
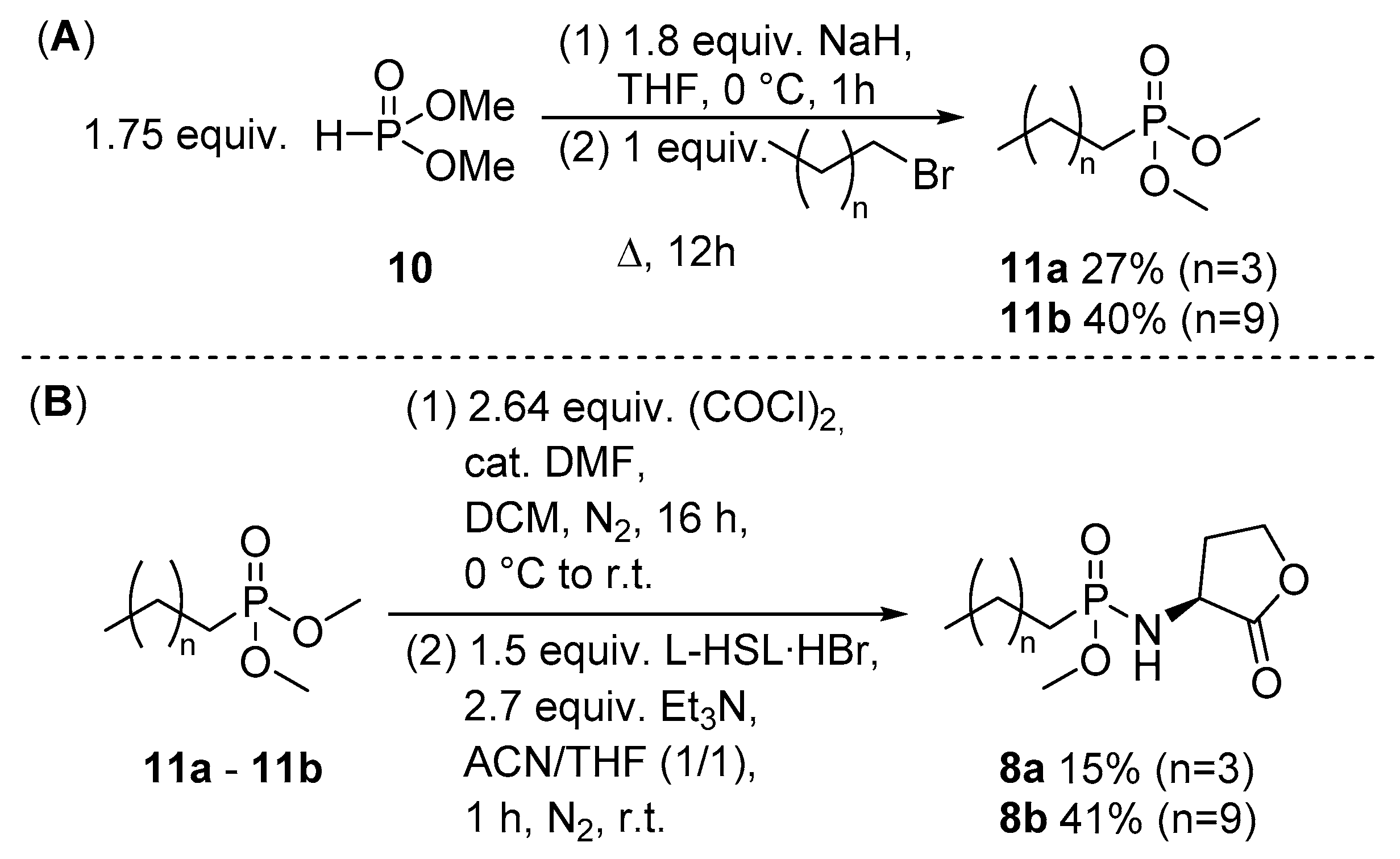
2.1. Synthesis of Novel Phosphonyl Moieties
2.2. Application for the Synthesis of N-acyl Homoserine Lactone Analogues
2.3. Application for the Use of Phosphoryl Chlorides
2.4. Screening for Herbicidal Activity
3. Materials and Methods
3.1. General Procedure for the Synthesis of Phosphorus-Containing Products 2, 3, 4, 6 and 7
3.2. Chlorophyll Fluorescence (Fv/Fm) Measurement
3.3. Plant Materials and Chemical Treatments
3.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.; Luo, W.; Wang, Y.; Li, Z.; Ma, X.; Peng, A. Efficient Synthesis of Phosphonamidates through One-Pot Sequential Reactions of Phosphonites with Iodine and Amines. Chem. A Eur. J. 2020, 26, 14474–14480. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, Y.; Tian, X.; Zhang, X.; Zhang, Y. An Efficient Pd & Pt-Catalyzed H/D Exchange Approach towards the Synthesis of Deuterium-Labeled Antiviral Prodrugs, Tenofovir Disoproxil Fumarate and Tenofovir Alafenamide. ChemistrySelect 2018, 3, 8724–8728. [Google Scholar] [CrossRef]
- Bessières, M.; Hervin, V.; Roy, V.; Chartier, A.; Snoeck, R.; Andrei, G.; Lohier, J.F.; Agrofoglio, L.A. Highly Convergent Synthesis and Antiviral Activity of (E)-but-2-Enyl Nucleoside Phosphonoamidates. Eur. J. Med. Chem. 2018, 146, 678–686. [Google Scholar] [CrossRef]
- Yang, K.W.; Feng, L.; Yang, S.K.; Aitha, M.; Lacuran, A.E.; Oelschlaeger, P.; Crowder, M.W. New β-Phospholactam as a Carbapenem Transition State Analog: Synthesis of a Broad-Spectrum Inhibitor of Metallo-β-Lactamases. Bioorg. Med. Chem. Lett. 2013, 23, 5855–5859. [Google Scholar] [CrossRef]
- Kadri, H.; Taher, T.E.; Xu, Q.; Sharif, M.; Ashby, E.; Bryan, R.T.; Willcox, B.E.; Mehellou, Y. Aryloxy Diester Phosphonamidate Prodrugs of Phosphoantigens (ProPAgens) as Potent Activators of Vγ9/Vδ2 T-Cell Immune Responses. J. Med. Chem. 2020, 63, 11258–11270. [Google Scholar] [CrossRef] [PubMed]
- Wone, D.W.G.; Rowley, J.A.; Garofalo, A.W.; Berkman, C.E. Optimizing Phenylethylphosphonamidates for the Inhibition of Prostate-Specific Membrane Antigen. Bioorg. Med. Chem. 2006, 14, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Bringley, D.A.; Calimsiz, S.; Garber, J.A.O.; Huynh, H.; Mohan, S.; Sarma, K.; Shen, J.; Curl, J.; Kwong, B.; et al. Synthesis of Rovafovir Etalafenamide (Part III): Evolution of the Synthetic Process to the Phosphonamidate Fragment. Org. Process. Res. Dev. 2021, 25, 1247–1262. [Google Scholar] [CrossRef]
- Luo, M.; Groaz, E.; De Jonghe, S.; Snoeck, R.; Andrei, G.; Herdewijn, P. Amidate Prodrugs of Cyclic 9-(S)-[3-Hydroxy-2-(Phosphonomethoxy)Propyl]Adenine with Potent Anti-Herpesvirus Activity. ACS Med. Chem. Lett. 2018, 9, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Fredriksen, K.A.; Amedjkouh, M. Investigation of Reactive Intermediates and Reaction Pathways in the Coupling Agent Mediated Phosphonamidation Reaction. Eur. J. Org. Chem. 2016, 2016, 474–482. [Google Scholar] [CrossRef]
- Lentini, N.A.; Foust, B.J.; Hsiao, C.-H.C.; Wiemer, A.J.; Wiemer, D.F. Phosphonamidate Prodrugs of a Butyrophilin Ligand Display Plasma Stability and Potent Vγ9 Vδ2 T Cell Stimulation. J. Med. Chem. 2018, 61, 8658–8669. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.; Mullins, N.D.; Balzarini, J.; Maguire, A.R. Synthesis and Evaluation of Prodrugs of α-Carboxy Nucleoside Phosphonates. J. Org. Chem. 2022, 87, 14793–14808. [Google Scholar] [CrossRef] [PubMed]
- Quintiliani, M.; Balzarini, J.; McGuigan, C. Design, Synthesis, and Biological Evaluation of C1–Phosphonamidate Analogues of 2-Deoxy-D-Ribose-1-Phosphate. Tetrahedron 2013, 69, 9111–9119. [Google Scholar] [CrossRef]
- Adler, P.; Pons, A.; Li, J.; Heider, J.; Brutiu, B.R.; Maulide, N. Chemoselective Activation of Diethyl Phosphonates: Modular Synthesis of Biologically Relevant Phosphonylated Scaffolds. Angew. Chem. Int. Ed. 2018, 57, 13330–13334. [Google Scholar] [CrossRef] [PubMed]
- Backx, S.; Dejaegere, A.; Simoens, A.; Van de Poel, J.; Krasowska, D.; Stevens, C.V.; Mangelinckx, S. Triethylamine-Mediated Transformation of Phosphonates into Phosphonamidates. Eur. J. Org. Chem. 2023, 26, e202300172. [Google Scholar] [CrossRef]
- Broadhurst, M.D.; Tsang, T.H. Alkylphosphonamidate Herbicides. U.S. Patent 5,205,852, 27 April 1993. [Google Scholar]
- Fañanás-Mastral, M.; Feringa, B.L. Copper-Catalyzed Synthesis of Mixed Alkyl Aryl Phosphonates. J. Am. Chem. Soc. 2014, 136, 9894–9897. [Google Scholar] [PubMed]
- Iorga, B.; Carmichael, D.; Savignac, P. Phosphonate–Phosphonochloridate Conversion. Comptes Rendus l’Académie Sci. Ser. IIC Chem. 2000, 3, 821–829. [Google Scholar] [CrossRef]
- Bonneure, E.; De Baets, A.; De Decker, S.; Van den Berge, K.; Clement, L.; Vyverman, W.; Mangelinckx, S. Altering the Sex Pheromone Cyclo(L-Pro-L-Pro) of the Diatom Seminavis Robusta towards a Chemical Probe. Int. J. Mol. Sci. 2021, 22, 1037. [Google Scholar] [CrossRef] [PubMed]
- Brotzel, F.; Kempf, B.; Singer, T.; Zipse, H.; Mayr, H. Nucleophilicities and Carbon Basicities of Pyridines. Chem. A Eur. J. 2007, 13, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Brotzel, F.; Ying, C.C.; Mayr, H. Nucleophilicities of Primary and Secondary Amines in Water. J. Org. Chem. 2007, 72, 3679–3688. [Google Scholar] [CrossRef]
- An, F.; Maji, B.; Min, E.; Ofial, A.R.; Mayr, H. Basicities and Nucleophilicities of Pyrrolidines and Imidazolidinones Used as Organocatalysts. J. Am. Chem. Soc. 2020, 142, 1526–1547. [Google Scholar] [CrossRef] [PubMed]
- Kanzian, T.; Nigst, T.A.; Maier, A.; Pichl, S.; Mayr, H. Nucleophilic Reactivities of Primary and Secondary Amines in Acetonitrile. Eur. J. Org. Chem. 2009, 2009, 6379–6385. [Google Scholar] [CrossRef]
- Kumara, M.N.; Nakahara, T.; Kobayashi, S.; Fujio, M.; Mishima, M. Nucleophilicities of Alcohols and Water in Acetonitrile Based on Reactivities of Benzhydrylium Ions. Bull. Chem. Soc. Jpn. 2018, 91, 523–530. [Google Scholar] [CrossRef]
- Ruysbergh, E.; Stevens, C.V.; De Kimpe, N.; Mangelinckx, S. Synthesis and Analysis of Stable Isotope-Labelled N-Acyl Homoserine Lactones. RSC Adv. 2016, 6, 73717–73730. [Google Scholar] [CrossRef]
- Castang, S.; Chantegrel, B.; Deshayes, C.; Dolmazon, R.; Gouet, P.; Haser, R.; Reverchon, S.; Nasser, W.; Hugouvieux-Cotte-Pattat, N.; Doutheau, A. N-Sulfonyl Homoserine Lactones as Antagonists of Bacterial Quorum Sensing. Bioorg. Med. Chem. Lett. 2004, 14, 5145–5149. [Google Scholar] [CrossRef]
- Frezza, M.; Castang, S.; Estephane, J.; Soulère, L.; Deshayes, C.; Chantegrel, B.; Nasser, W.; Queneau, Y.; Reverchon, S.; Doutheau, A. Synthesis and Biological Evaluation of Homoserine Lactone Derived Ureas as Antagonists of Bacterial Quorum Sensing. Bioorg. Med. Chem. 2006, 14, 4781–4791. [Google Scholar] [CrossRef]
- Frezza, M.; Soulère, L.; Reverchon, S.; Guiliani, N.; Jerez, C.; Queneau, Y.; Doutheau, A. Synthetic Homoserine Lactone-Derived Sulfonylureas as Inhibitors of Vibrio Fischeri Quorum Sensing Regulator. Bioorg. Med. Chem. 2008, 16, 3550–3556. [Google Scholar] [CrossRef]
- Zhang, Q.; Queneau, Y.; Soulère, L. Biological Evaluation and Docking Studies of New Carbamate, Thiocarbamate, and Hydrazide Analogues of Acyl Homoserine Lactones as Vibrio Fischeri-Quorum Sensing Modulators. Biomolecules 2020, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Boukraa, M.; Sabbah, M.; Soulère, L.; El Efrit, M.L.; Queneau, Y.; Doutheau, A. AHL-Dependent Quorum Sensing Inhibition: Synthesis and Biological Evaluation of α-(N-Alkyl-Carboxamide)-γ-Butyrolactones and α-(N-Alkyl-Sulfonamide)-γ-Butyrolactones. Bioorg. Med. Chem. Lett. 2011, 21, 6876–6879. [Google Scholar] [CrossRef] [PubMed]
- Persson, T.; Hansen, T.H.; Rasmussen, T.B.; Skindersø, M.E.; Givskov, M.; Nielsen, J. Rational Design and Synthesis of New Quorum-Sensing Inhibitors Derived from Acylated Homoserine Lactones and Natural Products from Garlic. Org. Biomol. Chem. 2005, 3, 253–262. [Google Scholar] [CrossRef]
- Procházková, E.; Filo, J.; Cigáň, M.; Baszczyňski, O. Sterically-Controlled Self-Immolation in Phosphoramidate Linkers Triggered by Light. Eur. J. Org. Chem. 2020, 2020, 897–906. [Google Scholar] [CrossRef]
- Addanki, H.R.; Vallabhaneni, M.R.; Chennamsetty, S.; Pullagura, P.; Sagurthi, S.R.; Pasupuleti, V.R. An in Silico ADMET, Molecular Docking Study and Microwave-Assisted Synthesis of New Phosphorylated Derivatives of Thiazolidinedione as Potential Anti-Diabetic Agents. Synth. Commun. 2022, 52, 300–315. [Google Scholar] [CrossRef]
- Procházková, E.; Navrátil, R.; Janeba, Z.; Roithová, J.; Baszczyňski, O. Reactive Cyclic Intermediates in the ProTide Prodrugs Activation: Trapping the Elusive Pentavalent Phosphorane. Org. Biomol. Chem. 2019, 17, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.N.; Reddy, C.D.; Reddy, V.K.; Kumar, K.A.; Reddy, C.S. Synthesis of Some Amino Acid Ester Linked Dioxathiaphosphocin/Dioxaphosphocin Derivatives. Synth. Commun. 2001, 31, 2929–2933. [Google Scholar] [CrossRef]
- Baker, N.R. Chlorophyll Fluorescence: A Probe of Photosynthesis In Vivo. Annu. Rev. Plant Biol. 2008, 59, 89–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Varanasi, V.; Perez-Jones, A. A Nondestructive Leaf-Disk Assay for Rapid Diagnosis of Weed Resistance to Multiple Herbicides. Weed Sci. 2021, 69, 274–283. [Google Scholar] [CrossRef]
- Desmedt, W.; Ameye, M.; Filipe, O.; De Waele, E.; Van Nieuwerburgh, F.; Deforce, D.; Van Meulebroek, L.; Vanhaecke, L.; Kyndt, T.; Höfte, M.; et al. Molecular Analysis of Broad-Spectrum Induced Resistance in Rice by the Green Leaf Volatile Z-3-Hexenyl Acetate. J. Exp. Bot. 2023, 74, 6804–6819. [Google Scholar] [CrossRef]
- De Zutter, N.; Ameye, M.; Debode, J.; De Tender, C.; Ommeslag, S.; Verwaeren, J.; Vermeir, P.; Audenaert, K.; De Gelder, L. Shifts in the Rhizobiome during Consecutive in Planta Enrichment for Phosphate-Solubilizing Bacteria Differentially Affect Maize P Status. Microb. Biotechnol. 2021, 14, 1594–1612. [Google Scholar] [CrossRef] [PubMed]
- Koehne, I.; Pietschnig, R. Synthesis of Geminal Bis- and Tetrakisphosphonate Ester Derivatives and Their Coordination Behavior towards Ca(II) Ions. Eur. J. Inorg. Chem. 2022, 2022, e202200194. [Google Scholar] [CrossRef]
- Nathanael, J.G.; White, J.M.; Richter, A.; Nuske, M.R.; Wille, U. Oxidative Damage of Proline Residues by Nitrate Radicals (NO3˙): A Kinetic and Product Study. Org. Biomol. Chem. 2020, 18, 6949–6957. [Google Scholar] [CrossRef] [PubMed]
- Angle, S.R.; Henry, R.M. Studies toward the Synthesis of (+)-Palustrine: The First Asymmetric Synthesis of (−)-Methyl Palustramate. J. Org. Chem. 1998, 63, 7490–7497. [Google Scholar] [CrossRef] [PubMed]
- Delorme, V.; Raux, B.; Puppo, R.; Leclaire, J.; Cavalier, J.F.; Marc, S.; Kamarajugadda, P.K.; Buono, G.; Fotiadu, F.; Canaan, S.; et al. Supported Inhibitor for Fishing Lipases in Complex Biological Media and Mass Spectrometry Identification. Biochimie 2014, 107, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Antczak, M.I.; Montchamp, J.-L. Mild Synthesis of Organophosphorus Compounds: Reaction of Phosphorus-Containing Carbenoids with Organoboranes. Org. Lett. 2008, 10, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Li, C.K.; Tao, Z.K.; Shoberu, A.; Zhang, W.; Zou, J.P. Copper-Catalyzed Cross-Coupling of Alkyl and Phosphorus Radicals for C(Sp3)-P Bond Formation. Org. Lett. 2022, 24, 6083–6087. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Tichý, T.; Veeravalli, V.; Lam, J.; Alt, J.; Wu, Y.; Tenora, L.; Majer, P.; Slusher, B.S.; Rais, R. Enhanced Oral Bioavailability of 2-(Phosphonomethyl)-Pentanedioic Acid (2-PMPA) from Its (5-Methyl-2-Oxo-1,3-Dioxol-4-Yl)Methyl (ODOL)-Based Prodrugs. Mol. Pharm. 2019, 16, 4292–4301. [Google Scholar] [CrossRef] [PubMed]
- Syrpas, M.; Ruysbergh, E.; Stevens, C.V.; De Kimpe, N.; Mangelinckx, S. Synthesis and Biological Evaluation of Novel N-α-Haloacylated Homoserine Lactones as Quorum Sensing Modulators. Beilstein J. Org. Chem. 2014, 10, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Syrpas, M.; Ruysbergh, E.; Blommaert, L.; Vanelslander, B.; Sabbe, K.; Vyverman, W.; De Kimpe, N.; Mangelinckx, S. Haloperoxidase Mediated Quorum Quenching by Nitzschia Cf Pellucida: Study of the Metabolization of N-Acyl Homoserine Lactones by a Benthic Diatom. Mar. Drugs 2014, 12, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [PubMed]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Backx, S.; Desmedt, W.; Dejaegere, A.; Simoens, A.; Van de Poel, J.; Krasowska, D.; Audenaert, K.; Stevens, C.V.; Mangelinckx, S. Synthesis of Mixed Phosphonate Esters and Amino Acid-Based Phosphonamidates, and Their Screening as Herbicides. Int. J. Mol. Sci. 2024, 25, 4739. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094739
Backx S, Desmedt W, Dejaegere A, Simoens A, Van de Poel J, Krasowska D, Audenaert K, Stevens CV, Mangelinckx S. Synthesis of Mixed Phosphonate Esters and Amino Acid-Based Phosphonamidates, and Their Screening as Herbicides. International Journal of Molecular Sciences. 2024; 25(9):4739. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094739
Chicago/Turabian StyleBackx, Simon, Willem Desmedt, Andreas Dejaegere, Andreas Simoens, Jef Van de Poel, Dorota Krasowska, Kris Audenaert, Christian V. Stevens, and Sven Mangelinckx. 2024. "Synthesis of Mixed Phosphonate Esters and Amino Acid-Based Phosphonamidates, and Their Screening as Herbicides" International Journal of Molecular Sciences 25, no. 9: 4739. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094739