1. Introduction
From its birth, optical microscopy has been an instrumental tool for the study of the living world. After surpassing the diffraction barrier, with the advent of the first nanoscope, macromolecular complexes could be explored in situ in their natural environment [
1,
2]. Among super-resolution technologies, single-molecule localization microscopy (SMLM) with PALM, STORM, PAINT, and MINFLUX [
3], up to their most powerful variants, i.e., RESI [
4] and MINSTED [
5], introduced a completely different approach to imaging. Unlike traditional fluorescence microscopy imaging, image formation is not simply achieved by the direct storage of emitting signals.
In SMLM, with appropriate probes and environmental conditions, only few isolated molecules stochastically undergo light emission at a certain time. The position of single emitters is instead calculated from the fitting of the corresponding Point Spread Function (PSF) with precision that depends on the number of collected photons. The process of localization is then iterated for thousands of frames to reveal the distribution of the fluorescent molecules in the field of view. The calculated coordinates for each molecule are then employed to generate a topographic map, reaching localization precision from tens to few nanometers in the case of the most powerful techniques. Historically, Photo-Activated Localization Microscopy (PALM) [
6] paved the way for the development of the most modern SMLM technologies. However, PALM obtains high spatial resolution thanks to photoactivation processes of selected fluorescent molecules, often requiring the genetic manipulation of the sample for the expression of photoactivatable proteins. Even if new photoactivatable and photoconvertible molecules are available nowadays for conjugation to antibodies, the localization process based on progressive photobleaching or photoconversion still call for relatively long observation times, thus limiting the potential of the technique. The introduction of a chemically active environment able to temporally modulate the photophysical properties of emitters is the base of stochastic optical reconstruction microscopy (STORM) [
7] that, with its derivative direct STORM (dSTORM) [
8], contributed to the diffusion of SMLM technologies in applied biomedical research. The ability to perform an on–off transition from an emission prone to a dark state together with the use of selected buffers imposes important restrictions on the number of simultaneously analyzable channels, while the photobleaching effects makes the evaluation of the fraction of fluorescent molecules really localized at the end of the process particularly difficult. DNA Point Accumulation for Imaging in Nanoscale Topography (DNA PAINT) [
9] removed some of these limitations by using the binding–unbinding transient immobilization of fluorescent DNA oligos to illuminate the biomolecules. DNA PAINT generally requires, with respect to STORM, longer exposure times to reach high localization precision but it has the enormous advantage of a limited influence of photobleaching and, as mentioned below, contributes to increasing the number of analyzable channels in a sample. MINFLUX [
10] and its recent sister technology, MINSTED [
5], adopt a reversed point of view in the localization of molecules. Instead of looking at the intensity peaks, these technologies search for emitters by beam scouting for a minimum number of emitted photons. Traditional SMLM obtains high-precision measurements by asking for high number of photons to the sample, in the MINFLUX framework, this requirement is satisfied by the light provided by the localizing doughnut-shaped laser beam, with a drastic cut in the duration of the measurement. However, the underlying technologies require complex and expensive hardware that strongly limits access to these microscopies in their infant phase for the application in the world of biomedical research.
Resolution Enhancement by Sequential Imaging (RESI) now provides a spatial resolution comparable to cryo-electron microscopy at the level of the nanometer by a novel DNA barcoding technique associated with multiple re-observations of the target structure with different tagging probes [
4]. RESI, like the other traditional SMLM techniques, maintains the advantage of low-complexity instrumentation, making this approach available thanks to the limited costs.
Nonetheless, we have not assisted yet with the expected wide distribution of these novel technologies across research laboratories. One of the possible reasons for this resides in the price to pay when pushing for the highest spatial resolution. Working beyond the diffraction barrier implies a limitation in the other performances of the optical microscope such as sensitivity, speed, content (number of simultaneously observed parameters), and statistical sampling.
Moreover, the potential of the microscope has been frequently limited by man-driven usage. Human control results in diminished objectivity, low throughput, and semi- or poorly quantitative measurements. Consequently, efforts in moving towards an intelligent automated microscope, able to choose from the optimal observation conditions, are strongly desired and continuously growing. We recently published a novel pipeline to perform image-cytometry analysis by a standard motorized fluorescence microscope, reaching statistical sampling in the order of thousands of cells and content of several parameters [
11]. Events can then be analyzed to quantitatively identify simple or complex target phenotypes, re-localized at the microscope stage and finally re-measured at the maximal resolution by 3D diffraction-limited imaging, confocal microscopy, or sub-diffraction analysis, by Structured Illumination or STORM super-resolution microscopy. At the same time, we proposed that correlative microscopy among different optical technologies can then be employed to remove the limitations in content typical of sub-diffraction microscopy, an approach that can be applied both to cells and tissues.
This pipeline was extended here by including single-molecule localization microscopy (SMLM) and sequential imaging. Many published papers successfully discuss the possibility of increasing the number of observed parameters by sequential immunostaining, reaching the visualization of up to ten channels with the resolution between 10 and 20 nanometers in the case of Exchange PAINT Microscopy [
9]. However, the conjugation of antibodies to DNA oligos or fluorochromes can present issues, making the approach not easily applicable for all the laboratories. Using commercially available reagents, we demonstrated the possibility of obtaining a four-parameter analysis employing sequential measurements re-aligned thanks to the identification of fiducial points.
SMLM microscopy has been mainly employed to reveal details of biological structures in situ, localizing molecules with precision ten times smaller than the diffraction barrier. However, the difficulties in combining the analysis of multiple parameters in the same sample limited, until now, its use in the analysis of molecular interactions and their intracellular localization. The present work demonstrates the possibility of obtaining high-content super-resolution data to reveal the mutual compartmentalization of different molecular species according to the actual function they are exerting.
We applied the developed tools to further characterize the interaction between p53 and 53BP1 molecules. 53BP1 has been originally isolated in vitro as a p53 binder in two-hybrid screening [
12]. After the discovery of its key role in the DNA Damage Recognition and Response (DDR) process [
13,
14,
15], its function in supporting the action of the guardian of the genome has been frequently forgotten. However, many published papers shed light on the novel functions of 53BP1, linking in vivo 53BP1 protein to the p53-driven transcriptional program that determines cell fate [
16,
17,
18,
19]. In recent work, we applied automated image cytometry and correlative super-resolution microscopy to study the spatio-temporal dynamics of p53-53BP1 interaction following severe DNA damage by Ionizing Radiation [
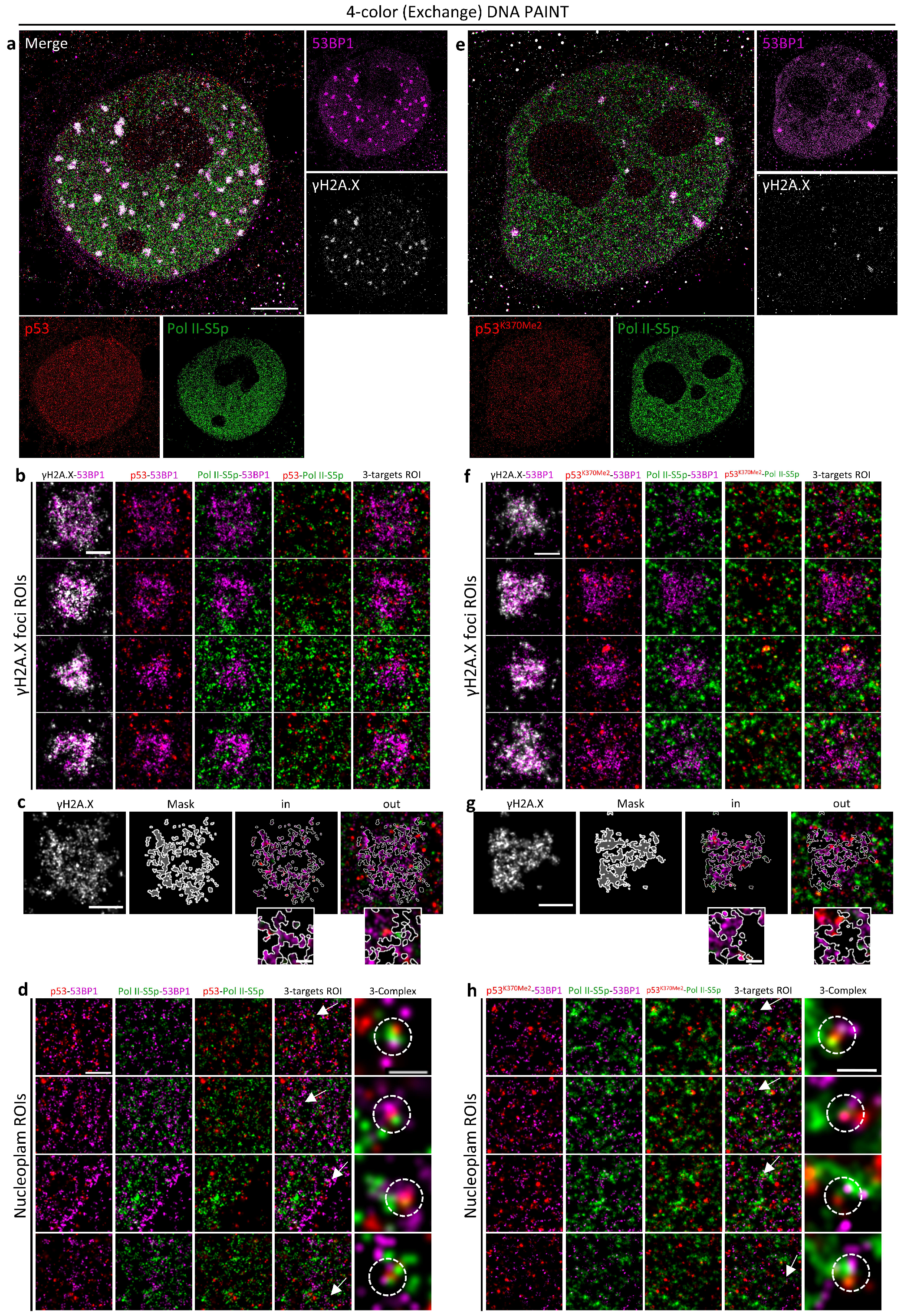
20]. Both spatial compartmentalization, with the formation of the putative p53-53BP1 complex out of the IR-induced γH2A.X foci, and temporal localization, with the detection of maximum interaction at late time points after irradiation when p53 was stabilized and damage foci reduced by the DNA-repair activity, suggested that 53BP1 can work to sustain the p53-controlled transcriptional program.
We thus focused on the molecular mechanisms at the base of the 53BP1 action in supporting the p53 transactivation function. Cells were classified according to (i) p53 and 53BP1 content, (ii) transcriptional activity by RNA Polymerase II phosphorylated on Serine 5 (Pol II-S5p), and (iii) DNA integrity by γH2A.X foci detection by diffraction-limited widefield microscopy and re-localized for super-resolution analysis. A two-step SMLM analysis was employed to monitor Pol II-S5p-p53, 53BP1-p53, Pol II-S5p-53BP1, and putative Pol II-S5p-53BP1-p53 interactions in and out of IR-induced foci, sustaining a model that includes 53BP1 in a putative complex with p53 as a transcription factor and basal transcription machinery. We also demonstrated that p53-53BP1 spatial compartmentalization can lead to the formation of putative complexes in the cytoplasm, thus suggesting the hypothesis that 53BP1 can also exert additional functions linked to its shuttling nature.
3. Discussion
Super-resolution microscopy revolutionized the world of optics by surpassing the diffraction barrier. This achievement was the result of considering the photophysical properties of the fluorescent molecules as active parts of the process, allowing the development of STED, PALM, STORM, and similar techniques that now provide a spatial resolution able to approach the performance of the electron microscope. However, different obstacles made it difficult for these novel technologies to spread into biomedical research laboratories. In some cases, the complexity of the required hardware and the consequent high costs strongly limit their availability for many potential interested users. Luckily, technological developments are rapidly contributing to the removal of this limiting aspect. However, some super-resolution techniques were built by relatively simple and accessible modifications of the fluorescence microscope.
Single-molecule localization microscopy (SMLM) reaches a resolution of tens of nanometers by just employing high-power laser sources and, eventually, specialized optics typical of Total Internal Reflection Microscopy. The large number of images required for the collection of a single sample unfortunately represents a big obstacle for “routine” use in applicative research. However, technology researchers are now paving the way for highly automated systems able to reach a throughput of thousands of analyzed cells [
35,
36].
On the other hand, classical SMLM approaches, i.e., PALM and STORM, suffer from a low number of simultaneously detectable fluorescence parameters due to their requirements regarding employable fluorochromes. DNA PAINT removed these limitations by sequential detection in Exchange PAINT microscopy [
9].
Even if these new tools are easily retrievable nowadays, SMLM, in most cases, has been mainly employed for single-parameter analysis to investigate in situ cellular structures at high resolution. Relatively few attempts have been made to investigate biomolecular interactions even after the reached increase in the achievable content, i.e., the number of measurable parameters on a single sample.
Correlative microscopy allows a dynamic choice of the real spatial resolution to be achieved in every step of an experiment flow (e.g., statistical sampling, duration of the experiment, content) [
11,
37,
38]. This way, all the performances of the microscope for the detection process can be optimized.
Here, we presented a pipeline to show the enormous potential of automated multimodal, multi-resolution correlative microscopy when working at a resolution in the order of tens of nanometers. Automated analysis-driven acquisition allows for the selection of quantitative and qualitative phenotypes producing both statistical reliability and high resolution. Adding an adapted Exchange PAINT protocol to this pipeline, we demonstrated how SMLM can be employed to (i) visualize the formation of putative macromolecular complexes and (ii) localize with precision in the order of 10–20 nanometers single molecules and complexes on intracellular structures.
The use of fluorescent nanodiamonds (FNDs) as sub-diffraction reference markers [
39] allowed us to modify the Exchange DNA PAINT protocol originally developed by Jungmann and coworkers to make the procedure accessible even in the absence of a microfluidic system on the microscope, simplifying the hardware requirements. Exchange PAINT is based on a series of sequential imaging phases: a set of DNA oligos conjugated to standard fluorochromes binds (and unbinds) to the corresponding target sequences linked to the employed primary antibodies, thus generating the single-molecule events. The first group of fluorescent oligos is then washed away by microfluidics and replaced by the injection of new fluorescent DNA sequences. The process is then iterated until all the required parameters are covered. In our protocol, the washing and reporter–oligo replacement were performed at the bench. Efficient repositioning was possible thanks to the use of an ad hoc designed sample holder and the employment of FND as reference markers (
Supplementary Figure S3).
We also propose an alternative approach for obtaining information at the spatial scale of tens of nanometers based on the combined use of proximity ligation analysis, diffraction-limited confocal microscopy, and SMLM. PLA visualizes putative macromolecular complexes measuring the proximity of the target molecules in the range of about 50 nm, a value close to the resolution reached by super-resolution microscopy techniques. Even if PLA efficiency is limited, its employment allows for (i) the obtainment of relevant statistical sampling thanks to the count of the number of interaction spots in a population composed by thousands of events, (ii) the targeting of the cells enriched by putative complexes, and (iii) their analysis for the interaction with additional candidates by correlative confocal-DNA PAINT and/or STORM microscopy. PLA may also function as a guide for post-acquisition data analysis by providing a reference region for the counting and clustering of events. Finally, PLA spots by themselves provide a useful marker for both drift correction, allowing for the maximization of the localization precision and the spatial registration of the images from different microscopy modalities, e.g., confocal versus SMLM, or from different SMLM imaging acquisitions as for Exchange DNA PAINT.
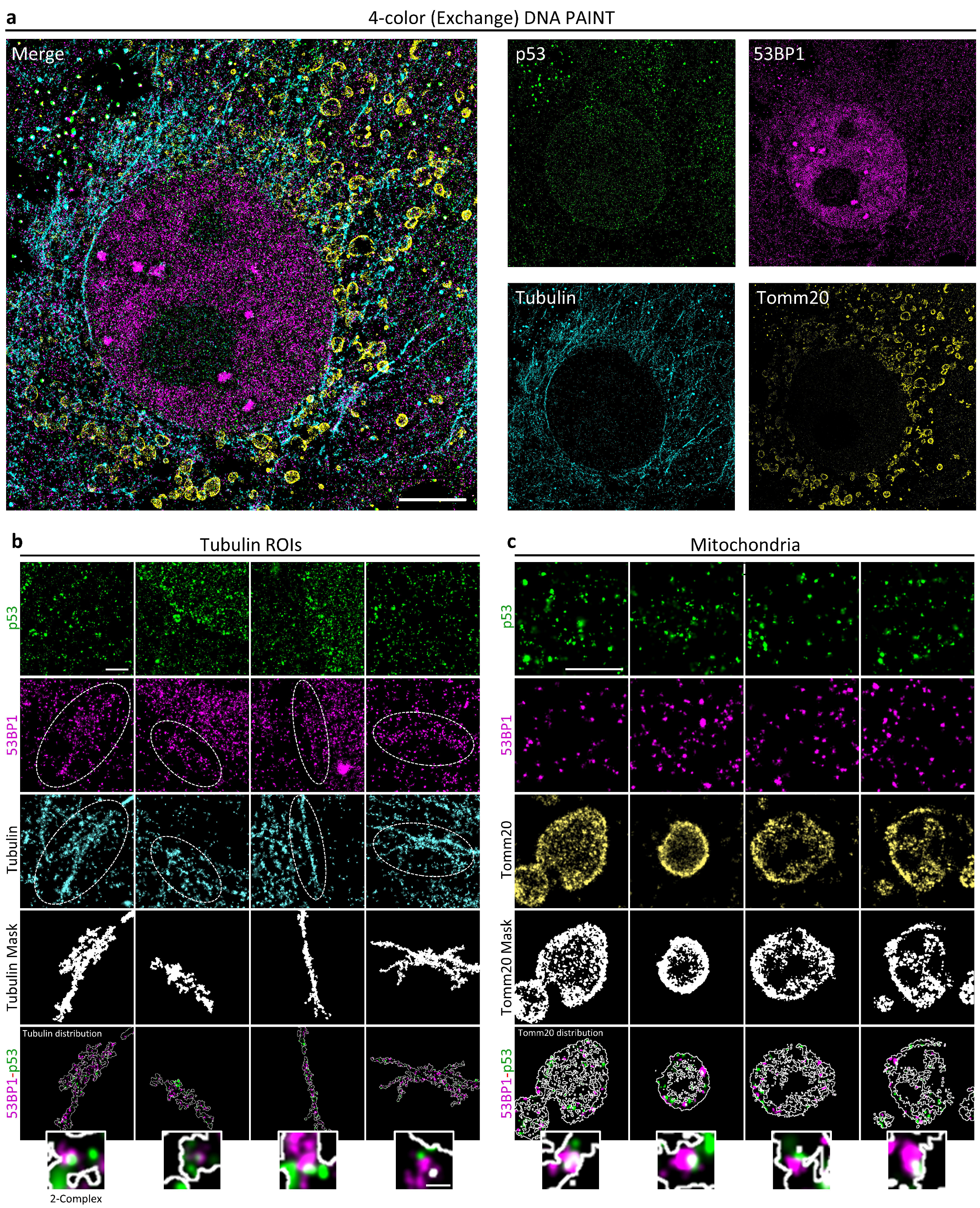
We validated the developed tools by analyzing the putative interaction between p53 and 53BP1 protein. As already mentioned, 53BP1 is simultaneously a major effector of DDR and a key interactor of the guardian of the genome to sustain its transcriptional program for cell-fate decisions. The employment of the new multimodal multiresolution and high-content SMLM pipeline allowed the detection and localization of p53-53BP1 molecular complexes with precision of less than 20 nm. Even if the literature already reported evidence of this interaction, this is the first time, to our knowledge, that the putative p53-53BP1 complex has been visualized in cells. The detection of 53BP1-p53-PolIIS5p putative complexes confirmed that 53BP1 transcriptionally related function occurs far from damaged DNA sites. High-content Exchange PAINT also provided clues on the existence of compartmentalization-regulated 53BP1-p53 interactions. 53BP1-p53 putative complexes have been detected in the cytoskeleton, providing an in situ confirmation of the potential role of 53BP1 in the regulation of p53 shuttling through the cell. We also found 53BP1, in association with p53 or alone, in the mitochondria. 53BP1’s function in the control of mitochondria homeostasis has been previously hypothesized [
34] based on the effects produced by the knock-out of the gene and without proving the presence of the protein in the compartment. This is the first proof, to our knowledge, of 53BP1’s presence in the mitochondria. These observations support a 53BP1-putative function as cargo for p53 and simultaneously suggest the existence of novel activities not necessarily related to p53.
Even if automated cytometry associated with multimodal correlative microscopy provides advantages in the reachable throughput of SMLM techniques, new developments are required to remove the limitations in their statistical sampling.
With this in mind, we are aware that the biological observations reported here may require further validation, considering their limited, even if increased, throughput, i.e., in the order of tens of cells. However, they can be seen as additional confirmation and refinements of already existing models. At the same time, we would like to underline that the main goal of the present work is to show the potential of the developed pipeline to favor the spreading of the SMLM technologies as a complement to the pure molecular biology approach.
Automated image cytometry coupled with multimodal high- and super-resolution microscopy can contribute to a new generation of fluorescence microscopy applications. The dynamic adaptation of the employed observation technique with the optimal spatial resolution and acquisition performances can have a profound impact on the management of experimental data. The automated selection of targets for high-resolution analysis in fixed samples enormously reduces storage efforts and makes the observation of rare phenotypes with increased statistical sampling possible [
40]. For example, when coupled to artificial intelligence, the approach can have a big impact in the contribution of microscopy to pathology [
41,
42,
43,
44]. In basic biomedical research, the presented methodologies are now gaining an increasing space with event-driven acquisitions [
40,
45,
46,
47]. The simultaneous development of new computational tools is now providing solutions to dramatically power up the statistical sampling in SMLM [
35,
36,
48], and the possibility of retrieving high-resolution sub-diffraction data from few acquired frames or even from a single snapshot [
49] can offer new solutions to measure molecular dynamics in real time. At the same time, the introduction of novel microscopy techniques in the framework of acquisition-driven image cytometry can generate a great opportunity to complement and bypass present limitations. Super-resolution microscopy and SMLM are now indeed able to reconstruct the distribution of fluorescent molecules with a sub-nanometer resolution. However, it is necessary to keep in mind that what we are gaining is a view of tagging fluorescent molecules in a sample; label-free methods are thus strongly required to gain a more precise understanding of the molecular mechanisms that rule life. Fluorescence Lifetime Microscopy (FLIM) provides one of the examples of how microscopy can evolve in this direction. The temporal resolution achieved, together with its increased optical penetration when coupled to multi-photon excitation, provides a tool able to monitor events in vivo at the molecular level without any exogenous fluorescence carrier that can easily fit with optimal performance in the general context of event-driven intravital microscopy [
50,
51,
52,
53,
54].
4. Materials and Methods
4.1. Cell Culture
MCF10A cells from the American Tissue Culture Collection (ATCC) were cultured in 50% DMEM High Glucose with stable L-glutamin (DMEM) (Euroclone) + 50% Ham’s F12 Medium (ThermoFisher Scientific, Waltham, MA, USA) containing 5% horse serum, 50 ng/mL penicillin/streptomycin (both from Euroclone, Milan, Italy), cholera toxin (Merck Life Science, Milan, Italy.), 10 mg/mL insulin (Merck Life Science, Milan, Italy), 500 ng/mL hydrocortisone (Merck Life Science, Milan, Italy), and 20 ng/mL EGF (Pepro Tech, Cranbury, NJ, USA,) at 37°C in 5% CO2. Cells were cultured on glass-bottom dishes (MatTek, Ashland, MA, USA). Growing cells were fixed for 10 min in 4% paraformaldehyde (wt/vol) to guarantee the exponential phase, whereas irradiated cells were exposed to 5-Gy radiation by an X-ray machine and fixed after 24 and 48 h.
4.2. Immunofluorescence of MCF10A Cells
Fixed MCF10A cells were washed and permeabilized for 10 min in a buffer containing 0.1% Triton X-100 (vol/vol) in PBS. EdU incorporation into DNA was detected following the manufacturer’s instructions by Biotin Azide (PEG4 carboxamide-6-Azidohexanyl Biotin) (ThermoFisher Scientific, Waltham, MA, USA) and Goat anti-biotin DyLight™ 800 (600145098, Rockland Immunochemicals, Pottstown, PA, USA). All the steps of the Click-iT reaction were performed at RT. After EdU detection, samples were incubated for 30 min in a blocking solution, 5% BSA (wt/vol) in PBS, and then overnight at 4 °C with primary antibodies in blocking solution. After washing in PBS, samples were incubated with secondary antibodies for 45 min. The following primary and secondary antibodies were employed: anti-53BP1 (ab36823, Abcam, Cambridge, UK) detected by Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Pacific Orange™ (ThermoFisher Scientific, Waltham, MA, USA), mouse anti-p53 Igg2a (sc-126, Santa Cruz Biotechnologies, Dallas, TX, USA) detected by Alexa Fluor® 488 AffiniPure™ Goat Anti-Mouse IgG, Fcγ subclass 2a specific (115-545-206, Jackson-immunoresearch, West Grove, PA, USA), rat anti-RNA Pol II Ser-5P (3e8-1 ChromoTek, Planegg, Germany) detected by Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 555 (ThermoFisher Scientific, Waltham, MA, USA), mouse anti-phosphoH2A.X (ser39) (γH2A.X) Igg1 (613402, Biolegend, San Diego, CA, USA) detected by Alexa Fluor® 647 AffiniPure™ Goat Anti-Mouse IgG, Fcγ subclass 1 specific (115-605-205, Jackson-immunoresearch, West Grove, PA, USA), Horizon™ V450 mouse anti-KI67 (561281, BD Biosciences, Franklin Lakes, NJ, USA). DNA was counterstained with Hoechst 33342, Trihydrochloride, and Trihydrate with a 10 mg/mL final concentration (ThermoFisher Scientific, Waltham, MA, USA). Finally, coverslips were then mounted in Slowfade Gold Antifade Mountant (ThermoFisher Scientific, Waltham, MA, USA).
4.3. In Situ Proximity Ligation Analysis (PLA)
Fixed MCF10A cells were washed and permeabilized for 10 min in a buffer containing 0.1% Triton X-100 (vol/vol) in PBS. Samples were then processed for an in situ proximity ligation assay (PLA) using NaveniFlex MR green detection reagent (Navinci, Uppsala, Sweden) according to the manufacturer’s instructions. The primary antibodies employed for PLA were rabbit anti-53BP1 (ab36823, Abcam, Cambridge, UK), mouse anti-p53 Igg2a (sc-126, Santa Cruz Biotechnologies, Dallas, TX, USA), rabbit Anti-Di-Methyl-TP53-Lys370 (STJ90115, St John’s Laboratory, London, UK), and mouse anti-53BP1 (612522, BD Biosciences, Franklin Lakes, NJ, USA). After the PLA reaction, the cells were processed for standard immunofluorescence for the detection of additional markers.
4.4. DNA PAINT Immunostaining
Sample were imaged by a commercial inverted Nikon Eclipse Ti2 microscope (Nikon instruments, Tokyo, Japan) equipped with an A1R confocal scan-head and N-SIM and N-STORM modules (Nikon instruments, Tokyo, Japan) and controlled by NIS Elements software (version 5.42.01). For widefield microscopy, an LED light source (pE-4000, CoolLED, Andover, United Kingdom) with 16 selectable wavelengths for widefield microscopy was employed. The light source for confocal imaging was a laser unit (LU-NV, Nikon instruments, Tokyo, Japan) equipped with 5 laser lines (405 nm (23.1 mW), 440 nm (25.5 mW) 488 nm (79.1 mW), 561 nm (79 mW), 647 nm (137 mW)), while a laser bench (L4Cc combiner, Oxxius S.A., Lannion, France) equipped with four high-power sources (405nm (216 mW), 488 nm (240 mW), 561 nm (240 mW), 640 nm (360 mW)) was employed for single-molecule localization experiments. A filter wheel (Optospin, Cairn Research Ltd, Faversham, Kent, UK) was placed in front of a CMOS camera (Dual ORCA Flash 4.0 Digital CMOS camera C13440, Hamamatsu, Tokyo, Japan) to acquire 16-bit scaled images (Widefield/DNA PAINT).
4.5. Microscope Setup
All data were acquired with a commercial inverted Nikon Eclipse Ti2 microscope (Nikon instruments, Tokyo, Japan), equipped with an A1R confocal scanhead and N-SIM and N-STORM modules (Nikon instruments, Tokyo, Japan). The fully motorized automated microscope was controlled by the NIS Elements software (version 5.42.01). The system performed multicolor widefield, confocal, and single-molecule localization imaging thanks to (i) an LED light source (pE-4000, CoolLED, Andover, UK) with 16 selectable wavelengths for widefield microscopy; (ii) a laser unit (LU-NV, Nikon instruments, Tokyo, Japan) equipped with 5 laser lines (405 nm (23.1 mW), 440 nm (25.5 mW) 488 nm (79.1 mW), 561 nm (79 mW), 647 nm (137 mW)) for confocal microscopy, and (iii) a laser bench (L4Cc combiner, Oxxius S.A., Lannion, France) equipped with four high-power sources (405 nm (216 mW), 488 nm (240 mW), 561 nm (240 mW), 640 nm (360 mW)) and two acousto-optic modulators for single-molecule microscopy). Emitted light was filtered by a filter wheel (Optospin, Cairn Research Ltd., Faversham, Kent, UK) and then was collected by a CMOS camera (Dual ORCA Flash 4.0 Digital CMOS camera C13440, Hamamatsu, Tokyo, Japan) set on a 16-bit scale detection modality (Widefield/DNA PAINT).
4.5.1. Widefield Microscopy
A 60x Plan Apo 1.4 NA objective was employed in widefield microscopy, obtaining diffraction-limited imaging in the order of the collected wavelengths. The exposure time per each fluorescence channel maximized the dynamic range, avoiding saturation. Optimal values were set in a preliminary observation of randomly chosen positions (between 30 and 200 ms per frame).
4.5.2. Confocal Microscopy
A Nikon A1R confocal microscope with a 100x 1.49 NA Apochromat objective (Nikon instruments, Tokyo, Japan) was employed to obtain confocal images. The employed conditions minimized crosstalk in favor of an optimal signal-to-noise ratio. During correlative confocal-DNA PAINT, channel acquisitions were sequentially performed with the high-speed galvanometric scanning mirrors (4x Line Average) to maximize image quality in a reduced time. The pinhole aperture was set to 0.6 Airy Unit. Data were collected with a digital size of 1024 × 1024 pixels, with a pixel of ∼0.065 μm. Scanning pixel dwell time (7 fps) and laser power were set to limit the photobleaching effect.
4.5.3. DNA PAINT Microscopy
Single-molecule imaging was performed using the system described in previous works [
11] using a Nikon CFI SR Apochromat TIRF 100× oil objective (1.49 NA). ATTO655 and Cy3b imagers were excited by using 561nm and 640nm laser wavelengths (L4Cc combiner, Oxxius S.A., Lannion, France). A multi-band dichroic mirror (C-NSTORM QUAD 405/488/561/647 FILTER SET; Chroma Technology Corporation, Bellows Falls, VT, USA), combined with 561 nm and 647 nm emission filters (Semrock Brightline
®, IDEX Health & Science, West Henrietta, NY, USA), was used to filter the fluorescence excitation. The acquisitions of 2 channels (correlative experiments) and 4 channels (Exchange PAINT) were performed sequentially. Fluorescent nanodiamonds (40 nm size) conjugated to streptavidin (Adamas Nanotechnologies, Raleigh, NC, USA), previously incubated with cells as described below, were excited by the 488 nm laser line and collected every 1000 frames to correct the drift between frames and the shift among channels.
Z drift was compensated in real time by a hardware autofocusing system based on the reflection of a near-infrared light (Perfect Focus System (PFS), Nikon instruments, Tokyo, Japan).
The number of frames and exposure time per channel depends on the density pattern of the immunostaining and imager concentration (at least 20,000 frames/channel with 90 ms of exposure time). Optimal imager concentrations were empirically determined according to the labeling density. The laser power was set according to the binding–unbinding rates of the imagers used. Additionally, the optimal imager concentration (and thus, the sufficiently low-fluorescence off-time) was necessary to ensure the proper number of binding events and thereby the robust detection of every docking strand during image acquisition. We employed 2–3 mL of imager buffer to maintain a constant concentration of imagers to minimize the negative effects of evaporation and photobleaching.
4.5.4. Exchange PAINT
For fluid exchange, a home-made clamping holder was constructed and mounted to the microscope stage, as shown in
Supplementary Figure S3. A schematic representation of the Exchange PAINT workflow can be found in
Supplementary Figure S4. Each image-acquisition step was followed with three brief washing steps (1 min) of washing buffer and one washing step of imaging buffer (1 min). Then, the next imager strand solution (imager + imaging buffer) was introduced. The acquisition and washing steps were repeated until all the four targets were imaged.
4.5.5. Drift-Correction Marker
Nitrogen-vacancy-center fluorescent nanodiamonds conjugated to streptavidin (FNDs; 40 nm; Adamas Nanotechnologies, Raleigh, NC, USA) were employed as reference markers for SMLM. Nanodiamonds stored in PBS with 0.1% BSA at 1 mg/mL (1% w/v) were diluted 1:50 and incubated with stained cells for 20’ at room temperature.
4.6. DNA PAINT Image Reconstruction
Single-molecule localization fitting was performed, with the Offline N-STORM Analysis module (NIS Elements software, version 5.42.01) correcting for spatial drift and chromatic aberrations. Before exporting DNA PAINT images, the Gaussian size of the single localizations and the format of the reconstructed images were set to optimize correlative imaging. We finally generated a dual-color DNA PAINT image of 41 × 41 μm (5 nm/px) with a Gaussian size of 10 nm. Localization precision was estimated in the range between 5 and 15 nm.
4.7. Automated Acquisition Protocol
The acquisition protocol was described in previous works [
11]. Briefly, a low-resolution map of the cells was obtained by a 4× objective, and regions of interest were chosen according to the optimal density and minimal presence of large cell aggregates. The selected ROIs were then acquired, and data were analyzed with the A.M.I.CO analysis package described below to select the phenotype of interest. The software recalculated stage coordinates, allowing repositioning for SMLM analysis.
4.8. A.M.I.CO Image Analysis
The A.M.I.CO analysis package was developed within the open-source ImageJ (version 1.54b) platform using its macro programming language, as described in detail in a previous study [
37]. The software is freely available upon request, or it can be downloaded from the GitHub public repository (
https://github.com/MarioFaretta/AMICO, accessed on 20 November 2022). Briefly, the software performs cell identification by automated segmentation providing integrated and mean values per pixel of all the acquired fluorescence channels. The measured data were then reorganized by an analysis module to perform statistical calculation to identify cell subpopulations.
4.9. Correlative Microscopy
Initially, a widefield image, centered on the cell of interest, was acquired in the channel employed for both DNA PAINT and confocal imaging. Spatial sampling was set to satisfy the Nyquist criterion (65 nm pixel size, no binning). The digital size of the collected image was adjusted to obtain a similar field of view (FOV) between WF and single-molecule images, but with different spatial sampling (65 nm for WF, 160 nm for DNA PAINT). DNA PAINT images were in fact acquired by inserting a 0.4x Relay lens to enlarge the FOV. A digital-size 256 × 256 pixel image in a DNA PAINT image matched with 620 × 620 pixels of a WF ROI (65 nm/pixel). WF images were also used to check the potential drift in the pictures collected by the single-molecule acquisition and the confocal image.
For confocal imaging, the pixel size was adjusted to match the spatial sampling of the WF image (about 65 nm). The pinhole size was set to 0.6 Airy unit. Bidirectional galvanometric scanning was enabled and the confocal acquisition parameters, such as line average, pixel dwell time, laser power, detector filters, and gain, were adjusted to increase quality signal without saturation while also minimizing background signal and photobleaching.
Then, the 2-color DNA PAINT acquisition was run. Single-molecule analysis on the two acquired datasets identified the positions of the fluorescent reporter molecules to reconstruct the two DNA PAINT images, including a correction of stage drift during the acquisition. Final alignment of the WF, confocal, and DNA PAINT images was performed after acquisition. The confocal and high-resolution WF images were aligned using registration software (the plugin included in NIS software version 5.42.01 or the open-source TurboReg plugin in the ImageJ platform were employed in this work). The same process was repeated to align the drifted widefield image, acquired with the 0.4x Relay lens of the single-molecule acquisition module. The registration parameters were finally applied to obtain the aligned reconstructed DNA PAINT images ([
11], Supplementary Protocol).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





