
Differential Solvent DEEP-STD NMR and MD Simulations Enable the Determinants of the Molecular Recognition of Heparin Oligosaccharides by Antithrombin to Be Disentangled

 , , and
, , and
Abstract
:1. Introduction
2. Results
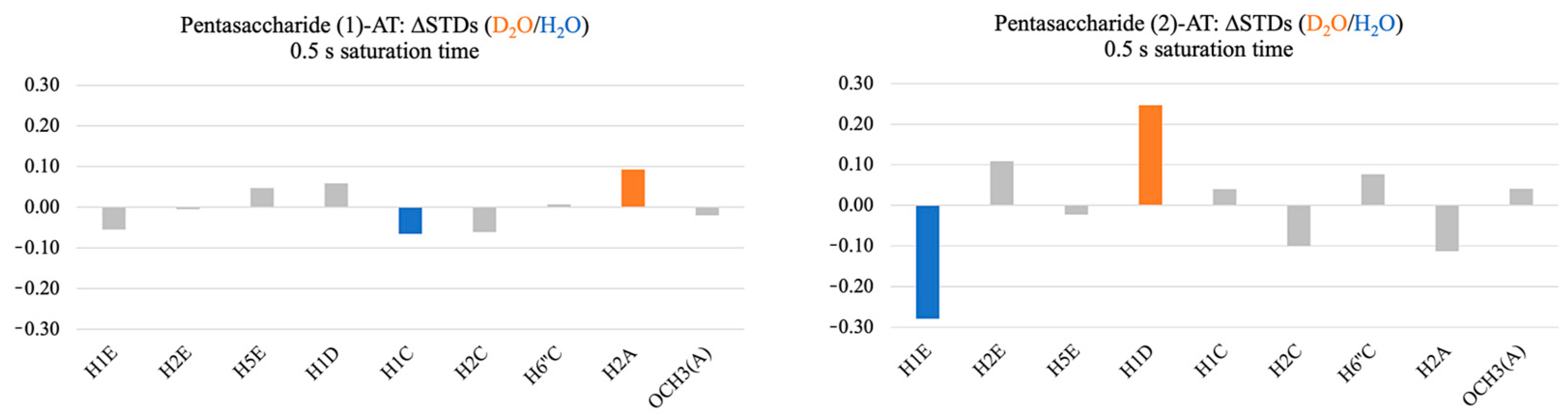
2.1. Comparison of the Interactions of Pentasaccharides (1) and (2) with AT Using STD and DEEP-STD NMR
2.2. Comparative Analysis of the Conformations of Pentasaccharides (1) and (2) in the Unbound and AT-Bound States by 3JH-H Coupling Constants and NOESY/tr-NOESY Experiments
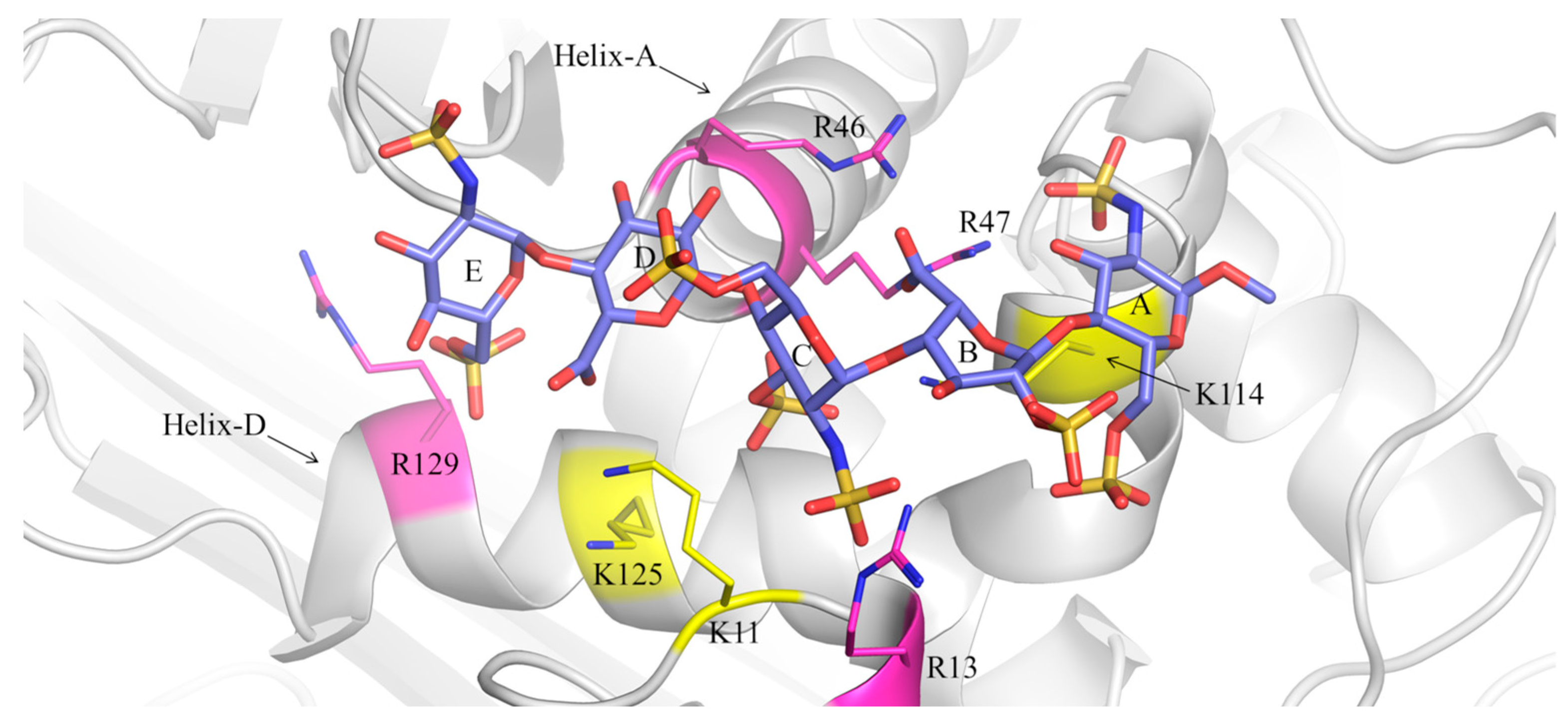
2.3. Comparison of the Interactions of Pentasaccharides (1) and (2) with AT by Molecular Docking
2.4. Investigation of the Structural Features of the Pentasaccharide (1)-AT and Pentasaccharide (2)-AT Complexes by MD Simulations
2.5. Comparison of the Solvation Properties at the Binding Interfaces of the Pentasaccharide (1)-AT and Pentasaccharide (2)-AT Complexes by MD Simulations
2.6. Model Validation by RedMat
3. Discussion
4. Materials and Methods
4.1. Ligands and Protein
4.2. NMR Experiments
4.3. Molecular Docking
4.4. Molecular Dynamics (MD) Simulations
4.5. Model Validation by RedMat
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lever, R.; Mulloy, B.; Page, C.P. (Eds.) Heparin—A Century of Progress; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2012; Volume 207. [Google Scholar]
- Lindahl, U.; Bäckström, G.; Thunberg, L.; Leder, I.G. Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proc. Natl. Acad. Sci. USA 1980, 77, 6551–6555. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Choay, J.; Ferro, D.R.; Gatti, G.; Jacquinet, J.C.; Petitou, M.; Provasoli, A.; Ragazzi, M.; Sinay, P.; Torri, G. Controversial glycosaminoglycan conformations. Nature 1986, 322, 215–216. [Google Scholar] [CrossRef]
- Sanderson, P.N.; Huckerby, T.N.; Nieduszynski, I.A. Conformational equilibria of α-L-iduronate residues in disaccharides derived from heparin. Biochem. J. 1987, 243, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Forster, M.J.; Jones, C.; Davies, D.B. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993, 293, 849–858. [Google Scholar] [CrossRef]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Casu, B.; Torri, G.; Bossennec, V.; Perly, B.; Sinaÿ, P.; Petitou, M.; Choay, J. Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences. Carbohydr. Res. 1990, 195, 157–167. [Google Scholar] [CrossRef]
- Björk, I.; Lindahl, U. Mechanism of the anticoagulant action of heparin. Mol. Cell. Biochem. 1982, 48, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Choay, J.; Petitou, M.; Lormeau, J.C.; Sinaÿ, P.; Casu, B.; Gatti, G. Structure-activity relationship in heparin: A synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochem. Biophys. Res. Commun. 1983, 116, 492–499. [Google Scholar] [CrossRef]
- Olson, S.T.; Björk, I.; Sheffer, R.; Craig, P.A.; Shore, J.D.; Choay, J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J. Biol. Chem. 1992, 267, 12528–12538. [Google Scholar] [CrossRef]
- Olson, S.T.; Richard, B.; Izaguirre, G.; Schedin-Weiss, S.; Gettins, P.G.W. Molecular mechanisms of antithrombin–heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie 2010, 92, 1587–1596. [Google Scholar] [CrossRef]
- Lindahl, U.; Hook, M. Glycosaminoglycans and Their Binding to Biological Macromolecules. Annu. Rev. Biochem. 1978, 47, 385–417. [Google Scholar] [CrossRef]
- Jackson, R.L.; Busch, S.J.; Cardin, A.D. Glycosaminoglycans: Molecular properties, protein interactions, and role in physiological processes. Physiol. Rev. 1991, 71, 481–539. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.P.; Sykes, B.D. The Two-Dimensional Transferred Nuclear Overhauser Effect: Theory and Practice. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 99–122. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Group Epitope Mapping by Saturation Transfer Difference NMR To Identify Segments of a Ligand in Direct Contact with a Protein Receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Peters, T. NMR Spectroscopy Techniques for Screening and Identifying Ligand Binding to Protein Receptors. Angew Chem. Int. Ed. Engl. 2003, 42, 864–890. [Google Scholar] [CrossRef] [PubMed]
- Monaco, S.; Tailford, L.E.; Juge, N.; Angulo, J. Differential Epitope Mapping by STD NMR Spectroscopy To Reveal the Nature of Protein-Ligand Contacts. Angew. Chem. Int. Ed. Engl. 2017, 56, 15289–15293. [Google Scholar] [CrossRef]
- Liepinsh, E.; Otting, G. Proton exchange rates from amino acid side chains—Implications for image contrast. Magn. Reson. Med. 1996, 35, 30–42. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef]
- Singh, A.; Tessier, M.B.; Pederson, K.; Wang, X.; Venot, A.P.; Boons, G.J.; Prestegard, J.H.; Woods, R.J. Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can. J. Chem. 2016, 94, 927–935. [Google Scholar] [CrossRef]
- Grootenhuis, P.D.; Boeckel, V. Constructing a molecular model of the interaction between Antithrombin-III and a potent synthetic heparin Analogue. J. Am. Chem. Soc. 1991, 113, 2743–2747. [Google Scholar] [CrossRef]
- Verli, H.; Guimarães, J.A. Insights into the induced fit mechanism in antithrombin–heparin interaction using molecular dynamics simulations. J. Mol. Graph. Model. 2005, 24, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.; Komáromi, I.; Bereczky, Z. The mechanism of high affinity pentasaccharide binding to antithrombin, insights from Gaussian accelerated molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 4718–4732. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Yu, W.; Desai, U.R.; MacKerell, A.D.; Mosier, P.D. Estimating glycosaminoglycan–protein interaction affinity: Water dominates the specific antithrombin–heparin interaction. Glycobiology 2016, 26, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Stancanelli, E.; Elli, S.; Hsieh, P.; Liu, J.; Guerrini, M. Recognition and Conformational Properties of an Alternative Antithrombin Binding Sequence Obtained by Chemoenzymatic Synthesis. ChemBioChem 2018, 19, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Elli, S.; Stancanelli, E.; Wang, Z.; Petitou, M.; Liu, J.; Guerrini, M. Degeneracy of the Antithrombin Binding Sequence in Heparin: 2-O-Sulfated Iduronic Acid Can Replace the Critical Glucuronic Acid. Chem. Eur. J. 2020, 26, 11814–11818. [Google Scholar] [CrossRef] [PubMed]
- Almond, A. Multiscale modeling of glycosaminoglycan structure and dynamics: Current methods and challenges. Curr. Opin. Struct. Biol. 2018, 50, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, K.K.; Sieradzan, A.K.; Samsonov, S.A. Molecular dynamics insights into protein-glycosaminoglycan systems from microsecond-scale simulations. Biopolymers 2019, 110, e23252. [Google Scholar] [CrossRef] [PubMed]
- Hricovíni, M.; Guerrini, M.; Bisio, A.; Torri, G.; Petitou, M.; Casu, B. Conformation of heparin pentasaccharide bound to antithrombin III. Biochem. J. 2001, 359, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Mourier, P.A.J.; Torri, G.; Viskov, C. Antithrombin-binding oligosaccharides: Structural diversities in a unique function? Glycoconj J. 2014, 31, 409–416. [Google Scholar] [CrossRef]
- Caldwell, E.E.O.; Nadkarni, V.D.; Fromm, J.R.; Linhardt, R.J.; Weiler, J.M. Importance of specific amino acids in protein binding sites for heparin and heparan sulfate. Int. J. Biochem. Cell Biol. 1996, 28, 203–216. [Google Scholar] [CrossRef]
- Fromm, J.R.; Hileman, R.E.; Caldwell, E.E.O.; Weiler, J.M.; Linhardt, R.J. Pattern and Spacing of Basic Amino Acids in Heparin Binding Sites. Arch. Biochem. Biophys. 1997, 343, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Thunberg, L.; Bäckström, G.; Riesenfeld, J.; Nordling, K.; Björk, I. Extension and structural variability of the antithrombin-binding sequence in heparin. J. Biol. Chem. 1984, 259, 12368–12376. [Google Scholar] [CrossRef]
- Craig, P.A.; Olson, S.T.; Shore, J.D. Transient kinetics of heparin-catalyzed protease inactivation by antithrombin III. Characterization of assembly, product formation, and heparin dissociation steps in the factor Xa reaction. J. Biol. Chem. 1989, 264, 5452–5461. [Google Scholar] [CrossRef] [PubMed]
- Petitou, M.; Casu, B.; Lindahl, U. 1976–1983, a critical period in the history of heparin: The discovery of the antithrombin binding site. Biochimie 2003, 85, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Petitou, M.; Lormeau, J.; Choay, J. Interaction of heparin and antithrombin III. Eur. J. Biochem. 1988, 176, 637–640. [Google Scholar] [CrossRef]
- Petitou, M.; van Boeckel, C.A.A. A Synthetic Antithrombin III Binding Pentasaccharide Is Now a Drug! What Comes Next? Angew. Chem. Int. Ed. Engl. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Pedersen, L.C. Anticoagulant heparan sulfate: Structural specificity and biosynthesis. Appl. Microbiol. Biotechnol. 2007, 74, 263–272. [Google Scholar]
- Jin, L.; Abrahams, J.P.; Skinner, R.; Petitou, M.; Pike, R.N.; Carrell, R.W. The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. USA 1997, 94, 14683–14688. [Google Scholar] [CrossRef]
- Desai, U.R.; Petitou, M.; Björk, I.; Olson, S.T. Mechanism of Heparin Activation of Antithrombin. J. Biol. Chem. 1998, 273, 7478–7487. [Google Scholar] [CrossRef]
- Petitou, M.; Barzu, T.; Herault, J.-P.; Herbert, J.-M. A unique trisaccharide sequence in heparin mediates the early step of antithrombin III activation. Glycobiology 1997, 7, 323–327. [Google Scholar] [CrossRef]
- Nepravishta, R.; Walpole, S.; Hicks, T.; Muñoz-García, J.C.; Angulo, J. Fast Validation of Static and Dynamic 3D Models of Weak Protein-Ligand Complexes from STD NMR Spectroscopy. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Mikhailov, D.; Mayo, K.H.; Vlahov, I.R.; Toida, T.; Pervin, A.; Linhardt, R.J. NMR solution conformation of heparin-derived tetrasaccharide. Biochem. J. 1996, 318, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Torri, G.; Casu, B.; Gatti, G.; Petitou, M.; Choay, J.; Jacquinet, J.C.; Sinaÿ, P. Mono- and bidimensional 500 MHz 1H-NMR spectra of a synthetic pentasaccharide corresponding to the binding sequence of heparin to antithrombin-III: Evidence for conformational peculiarity of the sulfated iduronate residue. Biochem. Biophys. Res. Commun. 1985, 128, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Torri, G.; Casu, B.; Gatti, G.; Jacquinet, J.C.; Sinay, P.; Petitou, M.; Choay, J. Evidence for conformational equilibrium of the sulfated L-iduronate residue in heparin and in synthetic heparin mono- and oligo-saccharides: NMR and force-field studies. J. Am. Chem. Soc. 1986, 108, 6773–6778. [Google Scholar] [CrossRef]
- Muñoz-García, J.C.; López-Prados, J.; Angulo, J.; Díaz-Contreras, I.; Reichardt, N.; de Paz, J.L.; Martín-Lomas, M.; Nieto, P.M. Effect of the substituents of the neighboring ring in the conformational equilibrium of iduronate in heparin-like trisaccharides. Chemistry 2012, 18, 16319–16331. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.H.; Thieker, D.F.; Guerrini, M.; Woods, R.J.; Liu, J. Uncovering the Relationship between Sulphation Patterns and Conformation of Iduronic Acid in Heparan Sulphate. Sci. Rep. 2016, 6, 29602. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Mallet, J.M.; Esnault, J.; Driguez, P.A.; Duchaussoy, P.; Sizun, P.; Hérault, J.P.; Herbert, J.M.; Petitou, M.; Sinaÿ, P. Synthesis of Conformationally Locked Carbohydrates: A Skew-Boat Conformation of L-Iduronic Acid Governs the Antithrombotic Activity of Heparin. Angew. Chem. Int. Ed. Engl. 2001, 40, 1670–1673. [Google Scholar] [CrossRef]
- Mosier, P.D.; Krishnasamy, C.; Kellogg, G.E.; Desai, U.R. On the Specificity of Heparin/Heparan Sulfate Binding to Proteins. Anion-Binding Sites on Antithrombin and Thrombin Are Fundamentally Different. PLoS ONE 2012, 7, e48632. [Google Scholar] [CrossRef] [PubMed]
- Elli, S.; Raffaini, G.; Guerrini, M.; Kosakovsky Pond, S.; Matrosovich, M. Molecular modeling and phylogenetic analyses highlight the role of amino acid 347 of the N1 subtype neuraminidase in influenza virus host range and interspecies adaptation. Front. Microbiol. 2023, 14, 1309156. [Google Scholar] [CrossRef]
- Petitou, M.; Jaurand, G.; Derrien, M.; Duchaussoy, P.; Choay, J. A new, highly potent, heparin-like pentasaccharide fragment containing a glucose residue instead of a glucosamine. Bioorg. Med. Chem. Lett. 1991, 1, 95–98. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. Knowl. Discov. Data Min. 1996, 96, 226–231. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Azzalini, A.; Menardi, G.; Rosolin, T. Cluster: Cluster Analysis via Nonparametric Density Estimation, version 1.0-0; Universitá di Padova: Padova, Italy, 2012. [Google Scholar]
- Weis, A.; Katebzadeh, K.; Soderhjelm, P.; Nilsson, I.; Ryde, U. Ligand affinities predicted with the MM/PBSA method: Dependence on the simulation method and the force field. J. Med. Chem. 2006, 49, 6596–6606. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.G.; Stone, J.E.; Kohlmeyer, A. Fast analysis of molecular dynamics trajectories with graphics processing units—Radial distribution function histogramming. J. Comput. Phys. 2011, 230, 3556–3569. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Raffaini, G.; Elli, S.; Ganazzoli, F. Computer simulation of bulk mechanical properties and surface hydration of biomaterials. J. Biomed. Mater. Res. A 2006, 77A, 618–626. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. Adsorption of charged albumin subdomains on a graphite surface. J. Biomed. Mater. Res. A 2006, 76A, 638–645. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | φ1/ψ1 | φ2/ψ2 | φ3/ψ3 | φ4/ψ4 |
|---|---|---|---|---|
| Pentasaccharide (1)-AT | −54°/−32° | 50°/9° | −61°/−46° | 59°/12° |
| Pentasaccharide (2)-AT | −41°/−29° | 43°/17° | −45°/−42° | 44°/6° (78%); −37°/−28° (22%) |
| Pentasaccharide-AT (PDB ID: 1AZX) | Pentasaccharide (1)-AT | Pentasaccharide (2)-AT | |
|---|---|---|---|
| Contacts | Dist. | Avg. Dist. | Avg. Dist. |
| GlcNS6S(E)(6S)-K125 | 6.0 | 4.9 | 6.1 |
| GlcNS6S(E)(6S)-R129 | 4.9 | 4.5 | 5.4 |
| GlcA(D)(COO−)-K125 | 4.9 | 7.3 | 4.5 |
| GlcNS3S6S(C)(NS)-R13 | 4.5 | 5.6 | 4.8 |
| GlcNS3S6S(C)(NS)-K114 | 3.8 | 5.1 | 4.1 |
| GlcNS3S6S(C)(6S)-R13 | 10.9 | 12.2 | 5.1 |
| GlcNS3S6S(C)(3S)-K114 | 3.1 | 3.9 | 4.9 |
| GlcNS3S6S(C)(6S)-R46 | 9.7 | 8.1 | 14.7 |
| IdoA2S(B)(2S)/ IdoA(B)(2OH)-R13 | 5.7 | 6.8 | 7.0 |
| IdoA2S(B)(COO−)/ IdoA(B)(COO−)-R46 | 5.1 | 5.5 | 10.6 |
| IdoA2S(B)(COO−) /IdoA(B)(COO−)-R47 | 5.0 | 4.4 | 6.8 |
| IdoA2S(B)(COO−)/ IdoA(B)(COO−)-K114 | 3.9 | 3.7 | 6.6 |
| GlcNS6S(A)(NS)-R46 | 6.8 | 7.4 | 11.4 |
| GlcNS6S(A)(NS)-R47 | 4.3 | 4.3 | 8.6 |
| GlcNS6S(A)(6S)-R13 | 4.7 | 7.3 | 11.2 |
| GlcNS6S(A)(6S)-K114 | 5.1 | 5.8 | 5.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parafioriti, M.; Elli, S.; Muñoz-García, J.C.; Ramírez-Cárdenas, J.; Yates, E.A.; Angulo, J.; Guerrini, M. Differential Solvent DEEP-STD NMR and MD Simulations Enable the Determinants of the Molecular Recognition of Heparin Oligosaccharides by Antithrombin to Be Disentangled. Int. J. Mol. Sci. 2024, 25, 4669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094669
Parafioriti M, Elli S, Muñoz-García JC, Ramírez-Cárdenas J, Yates EA, Angulo J, Guerrini M. Differential Solvent DEEP-STD NMR and MD Simulations Enable the Determinants of the Molecular Recognition of Heparin Oligosaccharides by Antithrombin to Be Disentangled. International Journal of Molecular Sciences. 2024; 25(9):4669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094669
Chicago/Turabian StyleParafioriti, Michela, Stefano Elli, Juan C. Muñoz-García, Jonathan Ramírez-Cárdenas, Edwin A. Yates, Jesús Angulo, and Marco Guerrini. 2024. "Differential Solvent DEEP-STD NMR and MD Simulations Enable the Determinants of the Molecular Recognition of Heparin Oligosaccharides by Antithrombin to Be Disentangled" International Journal of Molecular Sciences 25, no. 9: 4669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094669