
New Insights into the Genetic Basis of Lysine Accumulation in Rice Revealed by Multi-Model GWAS
Abstract
:1. Introduction
2. Results
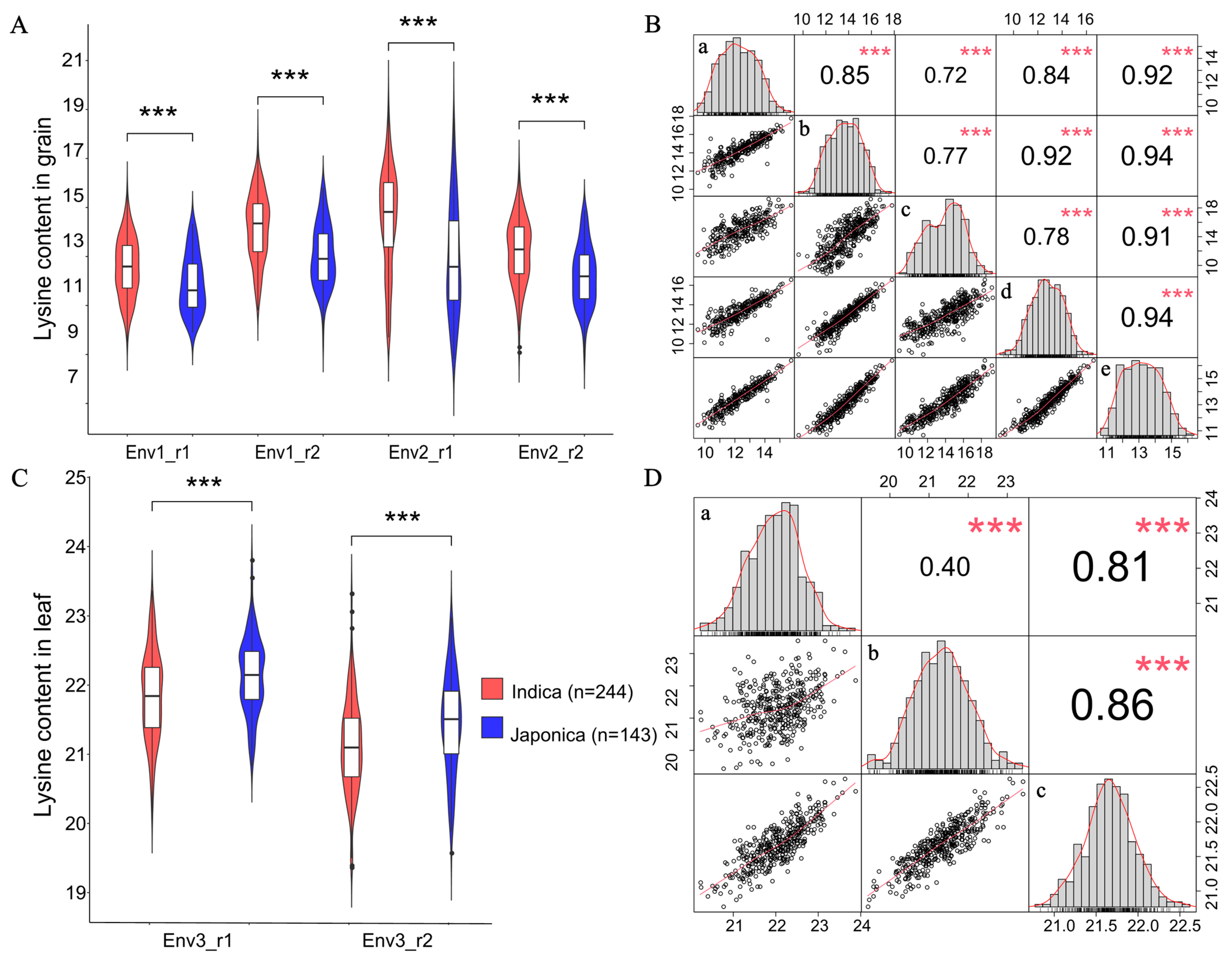
2.1. Lysine Content in Rice Grains and Leaves
2.2. Population Analysis
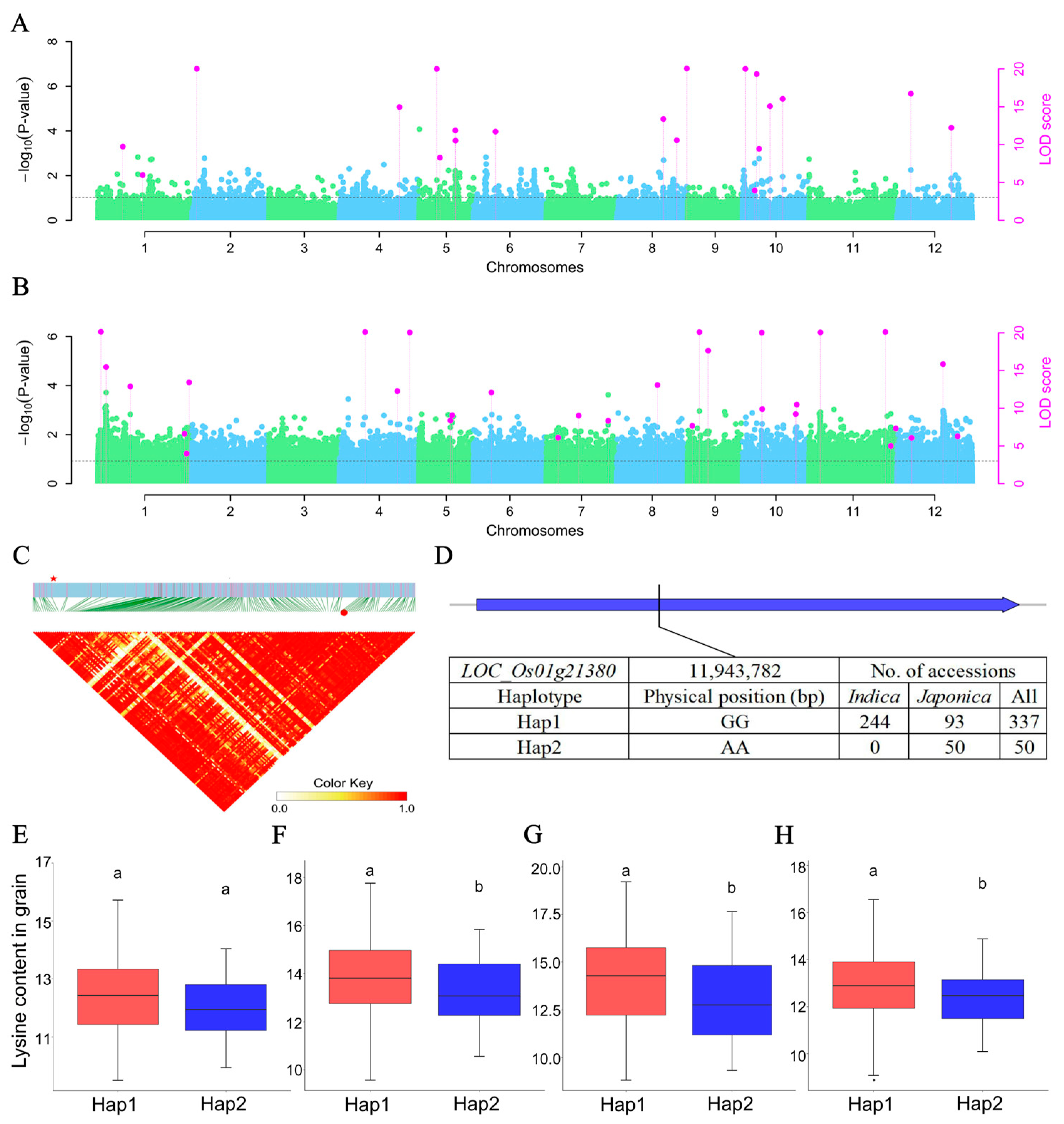
2.3. Identification and Application of QTNs Associated with Lysine Content
2.4. Candidate Genes for the Lysine Accumulation in Rice Grains
2.5. Candidate Genes for the Lysine Accumulation in Rice Leaves
2.6. Candidate Regulators Underlying the Lysine Accumulation in Rice Grains and Leaves
2.7. Lysine Content-Related QEI Detection and Candidate Genes
3. Discussion
3.1. Evaluation of QTNs Associated with Lysine Content in Rice
3.2. Candidate Genes Associated with Lysine Accumulation
3.3. Candidate Gene of Rice Lysine Accumulation Related QEI
3.4. Breeding Applications of Lysine Accumulation Associated QTNs and Genes
4. Materials and Methods
4.1. Plant Materials and Sample Sequencing
4.2. Metabolite Profiling
4.3. Population Structure and Linkage Disequilibrium Analysis
4.4. Genome-Wide Association Study
4.5. QTN Identification, Candidate Gene Analysis, and Genomic Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ufaz, S.; Galili, G. Improving the content of essential amino acids in crop plants: Goals and opportunities. Plant Physiol. 2008, 147, 954–961. [Google Scholar] [CrossRef]
- Galili, G.; Amir, R. Fortifying plants with the essential amino acids lysine and methionine to improve nutritional quality. Plant Biotechnol. J. 2013, 11, 211–222. [Google Scholar] [CrossRef]
- Galili, G.; Amir, R.; Fernie, A.R. The regulation of essential amino acid synthesis and accumulation in plants. Annu. Rev. Plant Biol. 2016, 67, 153–178. [Google Scholar] [CrossRef]
- Zhao, M.; Lin, Y.; Chen, H. Improving nutritional quality of rice for human health. Theor. Appl. Genet. 2020, 133, 1397–1413. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Rathore, R.; Singla, S.; Pareek, A. High lysine and high protein-containing salinity-tolerant rice grains (Oryza sativa cv IR64). Food Energy Secur. 2022, 11, e343. [Google Scholar] [CrossRef]
- Jin, C.; Fang, C.; Zhang, Y.; Fernie, A.R.; Luo, J. Plant metabolism paves the way for breeding crops with high nutritional value and stable yield. Sci. China Life Sci. 2021, 64, 2202–2205. [Google Scholar] [CrossRef]
- Shi, J.; An, G.; Weber, A.P.M.; Zhang, D. Prospects for rice in 2050. Plant Cell Environ. 2023, 46, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, N.K.; Ziska, L.H. Rice: Importance for Global Nutrition. J. Nutr. Sci. Vitaminol. 2019, 65, S2–S3. [Google Scholar] [CrossRef]
- Das, P.; Adak, S.; Lahiri Majumder, A. Genetic Manipulation for Improved Nutritional Quality in Rice. Front. Genet. 2020, 11, 776. [Google Scholar] [CrossRef]
- Wang, W.; Xu, M.; Wang, G.; Galili, G. New insights into the metabolism of aspartate-family amino acids in plant seeds. Plant Reprod. 2018, 31, 203–211. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Suen, P.K.; Zhang, C.Q.; Mak, W.S.; Gu, M.H.; Liu, Q.Q.; Sun, S.S. Improved growth performance, food efficiency, and lysine availability in growing rats fed with lysine-biofortified rice. Sci. Rep. 2017, 7, 1389. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, Y.; Jiang, Y. Amino Acids in Rice Grains and Their Regulation by Polyamines and Phytohormones. Plants 2022, 11, 1581. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Galili, G. Increased lysine synthesis coupled with a knockout of its catabolism synergistically boosts lysine content and also transregulates the metabolism of other amino acids in Arabidopsis seeds. Plant Cell 2003, 15, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Galili, G. Lysine metabolism is concurrently regulated by synthesis and catabolism in both reproductive and vegetative tissues. Plant Physiol. 2004, 135, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Kim, H.U.; Lee, Y.; Suh, S.C.; Lim, Y.P.; Lee, H.Y.; Kin, H.I. Constitutive and seed-specific expression of a maize lysine-feedback-insensitive dihydrodipicolinate synthase gene leads to increased free lysine levels in rice seeds. Mol. Breed. 2001, 8, 75–84. [Google Scholar] [CrossRef]
- Long, X.; Liu, Q.; Chan, M.; Wang, Q.; Sun, S.S. Metabolic engineering and profiling of rice with increased lysine. Plant Biotechnol. J. 2013, 11, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Frizzi, A.; Huang, S.; Gilbertson, L.A.; Armstrong, T.A.; Luethy, M.H.; Malvar, T.M. Modifying lysine biosynthesis and catabolism in corn with a single bifunctional expression/silencing transgene cassette. Plant Biotechnol. J. 2008, 6, 13–21. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Zhang, C.Q.; Chan, M.L.; Zhao, D.S.; Chen, J.Z.; Wang, Q.; Li, Q.F.; Yu, H.X.; Gu, M.H.; Sun, S.S.; et al. Biofortification of rice with the essential amino acid lysine: Molecular characterization, nutritional evaluation, and field performance. J. Exp. Bot. 2016, 67, 4285–4296. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gai, J. Genome-Wide Association Studies (GWAS). Methods Mol. Biol. 2023, 2638, 123–146. [Google Scholar] [CrossRef] [PubMed]
- Demirjian, C.; Vailleau, F.; Berthome, R.; Roux, F. Genome-wide association studies in plant pathosystems: Success or failure? Trends Plant Sci. 2023, 28, 471–485. [Google Scholar] [CrossRef]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.W.; Zhang, Z.C.; Xiang, Y.; Liu, M.H.; Zhou, Y.H.; Zuo, J.F.; Zhang, H.Q.; Chen, Y.; Zhang, Y.M. A compressed variance component mixed model for detecting QTNs, and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol. Plant 2022, 15, 630–650. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Feng, J.; Ren, W.; Huang, B.; Zhou, L.; Wen, Y.; Zhang, J.; Dunwell, J.; Xu, S.; Zhang, Y. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.; Ni, Y.; Zhang, Y. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zhang, H.; Ni, Y.; Huang, B.; Zhang, J.; Feng, J.; Wang, S.; Dunwell, J.; Zhang, Y.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Wen, Y.; Dunwell, J.; Zhang, Y. pKWmEB: Integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.; Ni, Y.; Wen, Y.; Niu, Y.; Tamba, C.; Yue, C.; Song, Q.; Zhang, Y. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Price, A.; Patterson, N.; Plenge, R.; Weinblatt, M.; Shadick, N.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.; McMullen, M.; Gaut, B.; Nielsen, D.; Holland, J.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef]
- Segura, V.; Vilhjalmsson, B.J.; Platt, A.; Korte, A.; Seren, U.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Jia, Z.; Dunwell, J.M. Editorial: The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits. Front. Plant Sci. 2019, 10, 100. [Google Scholar] [CrossRef]
- He, L.; Xiao, J.; Rashid, K.; Yao, Z.; Li, P.; Jia, G.; Wang, X.; Cloutier, S.; You, F. Genome-wide association studies for pasmo resistance in flax (Linum usitatissimum L.). Front. Plant Sci. 2018, 9, 1982–1997. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, H.; Sui, Y.; Miao, Y.; Jin, C.; Luo, J. Genome-wide association studies of five free amino acid levels in rice. Front. Plant Sci. 2022, 13, 1048860. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Che, Y.; Zhong, Y.; He, L. Genome-Wide Association Studies Using 3VmrMLM Model Provide New Insights into Branched-Chain Amino Acid Contents in Rice Grains. Plants 2023, 12, 2970. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sui, Y.; Che, Y.; Wang, H.; Rashid, K.Y.; Cloutier, S.; You, F.M. Genome-wide association studies using multi-models and multi-SNP datasets provide new insights into pasmo resistance in flax. Front. Plant Sci. 2023, 14, 1229457. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Fang, C.; Luo, J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. For. Cell Mol. Biol. 2019, 97, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Metabolite-based genome-wide association studies in plants. Curr. Opin. Plant Biol. 2015, 24, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, W.; Peng, M.; Gong, L.; Gao, Y.; Wan, J.; Wang, S.; Shi, L.; Zhou, B.; Li, Z.; et al. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 2016, 7, 12767. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shi, Y.; Liu, G.; Yao, F.; Zhang, Y.; Yang, C.; Guo, H.; Liu, X.; Jin, C.; Luo, J. Natural variation in the OsbZIP18 promoter contributes to branched-chain amino acid levels in rice. New Phytol. 2020, 228, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, Y.; Sun, Y.; Xie, Z.; Luo, Y.; Long, Q.; Feng, J.; Liu, X.; Wang, B.; He, D.; et al. Natural variations of OsAUX5, a target gene of OsWRKY78, control the contents of neutral essential amino acids in rice grains. Mol. Plant 2022, 16, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Fu, L.; Wang, B.; Ye, J.; Ou, W.; Yan, Y.; Li, M.; Zeng, L.; Dong, X.; Tie, W.; et al. Metabolic GWAS-based dissection of genetic basis underlying nutrient quality variation and domestication of cassava storage root. Genome Biol. 2023, 24, 289. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, B.; Qin, L.; Rahman, K.; Zhang, L.; Han, T. Transcription Factor: A Powerful Tool to Regulate Biosynthesis of Active Ingredients in Salvia miltiorrhiza. Front. Plant Sci. 2021, 12, 622011. [Google Scholar] [CrossRef]
- Zou, X.; Sun, H. DOF transcription factors: Specific regulators of plant biological processes. Front. Plant Sci. 2023, 14, 1044918. [Google Scholar] [CrossRef]
- Wen, B.; Luo, Y.; Liu, D.; Zhang, X.; Peng, Z.; Wang, K.; Li, J.; Huang, J.; Liu, Z. The R2R3-MYB transcription factor CsMYB73 negatively regulates l-Theanine biosynthesis in tea plants (Camellia sinensis L.). Plant Sci. 2020, 298, 110546. [Google Scholar] [CrossRef]
- Huang, Y.F.; Vialet, S.; Guiraud, J.L.; Torregrosa, L.; Bertrand, Y.; Cheynier, V.; This, P.; Terrier, N. A negative MYB regulator of proanthocyanidin accumulation, identified through expression quantitative locus mapping in the grape berry. New Phytol. 2014, 201, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, F.; Zhou, Y. The Application of Multi-Locus GWAS for the Detection of Salt-Tolerance Loci in Rice. Front. Plant Sci. 2018, 9, 1464. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Zhu, G.; Li, Y.; Li, W.; Fu, J.; Niu, E.; Li, L.; Zhang, D.; Guo, W. Genome-Wide Association Studies Reveal Genetic Variation and Candidate Genes of Drought Stress Related Traits in Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2018, 9, 1276. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, M.; Yan, Y.; Qing, C.; Zhang, X.; Zhang, Y.; Long, Y.; Wang, L.; Pan, L.; Zou, C.; et al. Genetic Dissection of Maize Embryonic Callus Regenerative Capacity Using Multi-Locus Genome-Wide Association Studies. Front. Plant Sci. 2018, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Alqudah, A.M.; Sallam, A.; Stephen Baenziger, P.; Borner, A. GWAS: Fast-forwarding gene identification and characterization in temperate Cereals: Lessons from Barley—A review. J. Adv. Res. 2020, 22, 119–135. [Google Scholar] [CrossRef]
- Sallam, A.; Martsch, R. Association mapping for frost tolerance using multi-parent advanced generation inter-cross (MAGIC) population in faba bean (Vicia faba L.). Genetica 2015, 143, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Bandillo, N.; Raghavan, C.; Muyco, P.A.; Sevilla, M.A.; Lobina, I.T.; Dilla-Ermita, C.J.; Tung, C.W.; McCouch, S.; Thomson, M.; Mauleon, R.; et al. Multi-parent advanced generation inter-cross (MAGIC) populations in rice: Progress and potential for genetics research and breeding. Rice 2013, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zheng, Z.; Fang, H.; Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 2021, 53, 1616–1621. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, H.; Zhang, Y.; Du, M.; Xiang, J.; Li, X.; Chang, Y.; Sun, J.; Cheng, X.; Xiong, M.; et al. Mining the candidate genes of rice panicle traits via a genome-wide association study. Front. Genet. 2023, 14, 1239550. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, R.; Ma, J.; Gao, H.; Deng, L.; Wang, N.; Wang, Y.; Zhang, J.; Li, K.; Zhang, W.; et al. Genome-wide association studies of yield-related traits in high-latitude japonica rice. BMC Genom. Data 2021, 22, 39. [Google Scholar] [CrossRef]
- Zhao, H.; Yao, W.; Ouyang, Y.; Yang, W.; Wang, G.; Lian, X.; Xing, Y.; Chen, L.; Xie, W. RiceVarMap: A comprehensive database of rice genomic variations. Nucleic Acids Res. 2015, 43, D1018–D1022. [Google Scholar] [CrossRef]
- Lu, K.; Wu, B.; Wang, J.; Zhu, W.; Nie, H.; Qian, J.; Huang, W.; Fang, Z. Blocking amino acid transporter OsAAP3 improves grain yield by promoting outgrowth buds and increasing tiller number in rice. Plant Biotechnol. J. 2018, 16, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Takaiwa, F. Differences in transcriptional regulatory mechanisms functioning for free lysine content and seed storage protein accumulation in rice grain. Plant Cell Physiol. 2010, 51, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yu, W.; Wu, H.; Zhang, C.; Sun, S.; Liu, Q. Lysine biofortification in rice by modulating feedback inhibition of aspartate kinase and dihydrodipicolinate synthase. Plant Biotechnol. J. 2021, 19, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.T.; Liu, E.; Polvi, S.L.; Pammett, R.T.; Page, J.E. A functional genomics screen identifies diverse transcription factors that regulate alkaloid biosynthesis in Nicotiana benthamiana. Plant J. 2010, 62, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liu, Y.; Luo, L.; Hussain, S.B.; Bai, Y.; Alam, S.M. Comparative Metabolites and Citrate-Degrading Enzymes Activities in Citrus Fruits Reveal the Role of Balance between ACL and Cyt-ACO in Metabolite Conversions. Plants 2020, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Tahjib-Ul-Arif, M.; Zahan, M.I.; Karim, M.M.; Imran, S.; Hunter, C.T.; Islam, M.S.; Mia, M.A.; Hannan, M.A.; Rhaman, M.S.; Hossain, M.A.; et al. Citric Acid-Mediated Abiotic Stress Tolerance in Plants. Int. J. Mol. Sci. 2021, 22, 7235. [Google Scholar] [CrossRef]
- Degu, A.; Hatew, B.; Nunes-Nesi, A.; Shlizerman, L.; Zur, N.; Katz, E.; Fernie, A.R.; Blumwald, E.; Sadka, A. Inhibition of aconitase in citrus fruit callus results in a metabolic shift towards amino acid biosynthesis. Planta 2011, 234, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.J.; Shah, J.K.; Brotman, Y.; Jahnke, K.; Willmitzer, L.; Kaiser, W.M.; Bauwe, H.; Igamberdiev, A.U. Inhibition of aconitase by nitric oxide leads to induction of the alternative oxidase and to a shift of metabolism towards biosynthesis of amino acids. J. Exp. Bot. 2012, 63, 1773–1784. [Google Scholar] [CrossRef]
- Goossens, J.; Mertens, J.; Goossens, A. Role and functioning of bHLH transcription factors in jasmonate signalling. J. Exp. Bot. 2017, 68, 1333–1347. [Google Scholar] [CrossRef]
- Hassani, D.; Fu, X.; Shen, Q.; Khalid, M.; Rose, J.K.C.; Tang, K. Parallel Transcriptional Regulation of Artemisinin and Flavonoid Biosynthesis. Trends Plant Sci. 2020, 25, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Lahham, M.; Jha, S.; Goj, D.; Macheroux, P.; Wallner, S. The family of sarcosine oxidases: Same reaction, different products. Arch. Biochem. Biophys. 2021, 704, 108868. [Google Scholar] [CrossRef] [PubMed]
- Goyer, A.; Johnson, T.L.; Olsen, L.J.; Collakova, E.; Shachar-Hill, Y.; Rhodes, D.; Hanson, A.D. Characterization and metabolic function of a peroxisomal sarcosine and pipecolate oxidase from Arabidopsis. J. Biol. Chem. 2004, 279, 16947–16953. [Google Scholar] [CrossRef]
- Reuber, B.E.; Karl, C.; Reimann, S.A.; Mihalik, S.J.; Dodt, G. Cloning and functional expression of a mammalian gene for a peroxisomal sarcosine oxidase. J. Biol. Chem. 1997, 272, 6766–6776. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, L.; Martin de Valmaseda, E.; Banuelos, O.; Lopez, P.; Riano, J.; Casqueiro, J.; Martin, J.F. Conversion of pipecolic acid into lysine in Penicillium chrysogenum requires pipecolate oxidase and saccharopine reductase: Characterization of the lys7 gene encoding saccharopine reductase. J. Bacteriol. 2001, 183, 7165–7172. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Nunes Nesi, A.; Araujo, W.L.; Braun, H.P. Amino Acid Catabolism in Plants. Mol. Plant 2015, 8, 1563–1579. [Google Scholar] [CrossRef]
- Araujo, W.L.; Ishizaki, K.; Nunes-Nesi, A.; Larson, T.R.; Tohge, T.; Krahnert, I.; Witt, S.; Obata, T.; Schauer, N.; Graham, I.A.; et al. Identification of the 2-hydroxyglutarate and isovaleryl-CoA dehydrogenases as alternative electron donors linking lysine catabolism to the electron transport chain of Arabidopsis mitochondria. Plant Cell 2010, 22, 1549–1563. [Google Scholar] [CrossRef]
- Leandro, J.; Houten, S.M. The lysine degradation pathway: Subcellular compartmentalization and enzyme deficiencies. Mol. Genet. Metab. 2020, 131, 14–22. [Google Scholar] [CrossRef]
- Biazzi, E.; Nazzicari, N.; Pecetti, L.; Brummer, E.C.; Palmonari, A.; Tava, A.; Annicchiarico, P. Genome-Wide Association Mapping and Genomic Selection for Alfalfa (Medicago sativa) Forage Quality Traits. PLoS ONE 2017, 12, e0169234. [Google Scholar] [CrossRef] [PubMed]
- Rakotondramanana, M.; Tanaka, R.; Pariasca-Tanaka, J.; Stangoulis, J.; Grenier, C.; Wissuwa, M. Genomic prediction of zinc-biofortification potential in rice gene bank accessions. Theor. Appl. Genet. 2022, 135, 2265–2278. [Google Scholar] [CrossRef]
- Qin, J.; Shi, A.; Song, Q.; Li, S.; Wang, F.; Cao, Y.; Ravelombola, W.; Song, Q.; Yang, C.; Zhang, M. Genome Wide Association Study and Genomic Selection of Amino Acid Concentrations in Soybean Seeds. Front. Plant Sci. 2019, 10, 1445–1460. [Google Scholar] [CrossRef] [PubMed]
- Singer, W.M.; Shea, Z.; Yu, D.; Huang, H.; Mian, M.A.R.; Shang, C.; Rosso, M.L.; Song, Q.J.; Zhang, B. Genome-Wide Association Study and Genomic Selection for Proteinogenic Methionine in Soybean Seeds. Front. Plant Sci. 2022, 13, 859109. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Brown, B.B.; Song, Q.; Vaughn, J.N.; Li, Z. Genomic Selection for Yield and Seed Composition Traits within an Applied Soybean Breeding Program. G3 2019, 9, 2253–2265. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Peterson, D.; Tamura, K. MEGA-CC: Computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 2012, 28, 2685–2686. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Dong, S.S.; He, W.M.; Ji, J.J.; Zhang, C.; Guo, Y.; Yang, T.L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Tamba, C.L.; Wen, Y.J.; Li, P.; Ren, W.L.; Ni, Y.L.; Gao, J.; Zhang, Y.M. mrMLM v4.0.2: An R Platform for Multi-locus Genome-wide Association Studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.W.; Xiang, Y.; Liu, M.H.; Zhang, Y.M. IIIVmrMLM: The R and C++ tools associated with 3VmrMLM, a comprehensive GWAS method for dissecting quantitative traits. Mol. Plant 2022, 15, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, Z.; Gao, J.; Wang, X.; Guo, K. CandiHap: A haplotype analysis toolkit for natural variation study. Mol. Breed. 2023, 43, 21. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Shen, S.; Zhou, S.; Li, Y.; Mao, Y.; Zhou, J.; Shi, Y.; An, L.; Zhou, Q.; Peng, W.; et al. Rice metabolic regulatory network spanning the entire life cycle. Mol. Plant 2022, 15, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Wang, K.; Abid, M.A.; Rasheed, A.; Crossa, J.; Hearne, S.; Li, H. DNNGP, a deep neural network-based method for genomic prediction using multi-omics data in plants. Mol. Plant 2023, 16, 279–293. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Number | Range | Mean | SD | Variance | Skewness | Kurtosis | CV (%) a | H2 |
|---|---|---|---|---|---|---|---|---|---|
| Grain_env1_r1 | 272 | 6.22 | 12.35 | 1.24 | 1.54 | 0.10 | −0.74 | 93.59 | 0.69 |
| Grain_env1_r2 | 364 | 8.19 | 13.75 | 1.44 | 2.08 | −0.03 | −0.63 | 107.49 | |
| Grain_env2_r1 | 365 | 10.41 | 13.90 | 2.29 | 5.26 | −0.18 | −0.82 | 165.98 | |
| Grain_env2_r2 | 365 | 7.68 | 12.82 | 1.38 | 1.91 | −0.09 | −0.40 | 102.84 | |
| Leaf_env3_r1 | 387 | 3.65 | 22.02 | 0.62 | 0.39 | −0.14 | −0.12 | 43.53 | 0.16 |
| Leaf_env3_r2 | 387 | 3.96 | 21.31 | 0.70 | 0.49 | 0.07 | −0.01 | 52.62 |
| Dataset | No. of Detected Common QTNs | R2 (%) | ||||
|---|---|---|---|---|---|---|
| GLM|MLM-SL | GLM|mrMLM-ML | MLM-SL|mrMLM-ML | GLM|MLM-SL|mrMLM-ML | Total | ||
| Grain_env1_r1 | 5 | 11 | 4 | 3 | 23 | 0.83–20.25 |
| Grain_env1_r2 | 21 | 26 | 5 | 3 | 55 | 0.12–25.82 |
| Grain_env2_r1 | 50 | 12 | 4 | 2 | 68 | 0.03–24.44 |
| Grain_env2_r2 | 7 | 27 | 3 | 1 | 38 | 0.05–26.08 |
| Grain_BLUP | 81 | 26 | 4 | 6 | 117 | 0.16–27.65 |
| Leaf_env3_r1 | 5 | 17 | 3 | 3 | 28 | 0.03–16.18 |
| Leaf_env3_r2 | 16 | 10 | 3 | 1 | 30 | 0.27–12.21 |
| Leaf_BLUP | 4 | 11 | 2 | 2 | 19 | 0.03–13.45 |
| Dataset | h2 | RRBLUP-r |
|---|---|---|
| Grain_env1_r1 | 0.54 | 0.76 |
| Grain_env1_r2 | 0.62 | 0.84 |
| Grain_env2_r1 | 0.56 | 0.76 |
| Grain_env2_r2 | 0.62 | 0.83 |
| Grain_BLUP | 0.64 | 0.85 |
| Leaf_env3_r1 | 0.30 | 0.71 |
| Leaf_env3_r2 | 0.30 | 0.65 |
| Leaf_BLUP | 0.34 | 0.77 |
| Common QTN | Gene Id | KEGG Pathway/Annotation | Functional Annotation | E-Value |
|---|---|---|---|---|
| QTN-sf0711949886 | LOC_Os07g20544 | Lysine biosynthesis | Aspartokinase | 5.9 × 10−181 |
| QTN-sf0906935953 | LOC_Os09g12290 | Lysine biosynthesis | Bifunctional aspartokinase/homoserine dehydrogenase | 0 |
| QTN-sf0103080436 | LOC_Os01g06600 | Lysine degradation | Glutaryl-CoA dehydrogenase | 8.5 × 10−152 |
| QTN-sf1012964749 | LOC_Os10g25130 | Alanine, aspartate, and glutamate metabolism | Aminotransferase | 6 × 10−247 |
| QTN-sf1012964749 | LOC_Os10g25140 | Alanine, aspartate, and glutamate metabolism | Aminotransferase | 1.7 × 10−214 |
| QTN-sf0311302595 | LOC_Os03g19930 | Alanine, aspartate, and glutamate metabolism | Adenylosuccinate lyase | 3.6 × 10−187 |
| QTN-sf0717867262 | LOC_Os07g30170 | Beta-Alanine metabolism | Nitrilase | 1.4 × 10−222 |
| QTN-sf0825353310 | LOC_Os08g40110 | Biosynthesis of amino acids | Peptidase | 1.1 × 10−149 |
| QTN-sf1013407412 | LOC_Os10g26010 | Biosynthesis of amino acids | Cystathionine gamma-synthase | 1.9 × 10−158 |
| QTN-sf0419067736 | LOC_Os04g31960 | Biosynthesis of amino acids | Thiamine pyrophosphate enzyme | 5.9 × 10−204 |
| QTN-sf0419067736 | LOC_Os04g32010 | Biosynthesis of amino acids | Thiamine pyrophosphate enzyme | 1.2 × 10−233 |
| QTN-sf0100906859 | LOC_Os01g02880 | Biosynthesis of amino acids | Fructose-bisphosphate aldolase isozyme | 9.3 × 10−195 |
| QTN-sf0110799569 | LOC_Os01g19220 | Cyanoamino acid metabolism | Beta-D-xylosidase | 1.7 × 10−304 |
| QTN-sf0607725091 | LOC_Os06g13820 | Cysteine and methionine metabolism | Dynamin, putative | 0 |
| QTN-sf0803340682 | LOC_Os08g06100 | Tryptophan metabolism | O-methyltransferase | 3.5 × 10−199 |
| QTN-sf0105539291 | LOC_Os01g10504 | Transcription factor | MADS-box family gene with MIKCc type-box | 1 × 10−95 |
| QTN-sf0308698430 | LOC_Os03g15660 | Transcription factor | AP2 domain-containing protein | 1.7 × 10−35 |
| QTN-sf0606188796 | LOC_Os06g11780 | Transcription factor | MYB family transcription factor | 4 × 10−80 |
| QTN-sf0626549077 | LOC_Os06g44010 | Transcription factor | Superfamily of TFs having WRKY and zinc finger domains | NA |
| QTN-sf0703936507 | LOC_Os07g07974 | Transcription factor | Tesmin/TSO1-like CXC domain-containing protein | 1.9 × 10−78 |
| QTN-sf1219521482 | LOC_Os12g32250 | Transcription factor | WRKY DNA-binding domain containing protein | NA |
| QTN-sf0336203804 | LOC_Os03g64260 | Transcription factor | AP2 domain-containing protein | 2.1 × 10−78 |
| QTN-sf0101545236 | LOC_Os01g03720 | Transcription factor | MYB family transcription factor | 8.7 × 10−68 |
| Common QTN | Gene Id | KEGG Pathway/Annotation | Functional Annotation | E-Value |
|---|---|---|---|---|
| QTN-sf0140574604 | LOC_Os01g70220 | Lysine degradation | Histone-lysine N-methyltransferase | 1.9 × 10−121 |
| QTN-sf1119083279 | LOC_Os11g33240 | Biosynthesis of amino acids | Citrate synthase | 2.1 × 10−140 |
| QTN-sf0140574604 | LOC_Os01g70170 | Alanine, aspartate, and glutamate metabolism | Transaldolase | 2 × 10−83 |
| QTN-sf0300274740 | LOC_Os03g01600 | Alanine, aspartate, and glutamate metabolism | Aminotransferase domain-containing protein | 3.2 × 10−147 |
| QTN-sf0314034319 | LOC_Os03g24460 | Alanine, aspartate, and glutamate metabolism | Aminotransferase domain-containing protein | 9 × 10−57 |
| QTN-sf0822892970 | LOC_Os08g36320 | Alanine, aspartate, and glutamate metabolism | Decarboxylase | 2.5 × 10−115 |
| QTN-sf0200325193 | LOC_Os02g01510 | Cysteine and methionine metabolism | Lactate/malate dehydrogenase | 2 × 10−156 |
| QTN-sf0111240543 | LOC_Os01g19970 | Transcription factor | MYB family transcription factor | 1.3 × 10−76 |
| QTN-sf0103404473 | LOC_Os01g07120 | Transcription factor | AP2 domain-containing protein | 4.5 × 10−40 |
| QTN-sf0135366231 | LOC_Os01g60960 | Transcription factor | DUF260 domain-containing protein | NA |
| QTN-sf0603336542 | LOC_Os06g06900 | Transcription factor | Helix-loop-helix DNA-binding domain-containing protein | NA |
| QTN-sf0702729577 | LOC_Os07g05720 | Transcription factor | TCP family transcription factor | NA |
| Dataset | QEI | Gene Id | KEGG Pathway | Functional Annotation | E-Value |
|---|---|---|---|---|---|
| Lys_grain | QEI-sf0111954416 | LOC_Os01g21380 | Lysine degradation | FAD-dependent oxidoreductase domain-containing protein | 3.2 × 10−115 |
| Lys_grain | QEI-sf0519512601 | LOC_Os05g33380 | Biosynthesis of amino acids | Fructose-bisphosphate aldolase isozyme | 3.5 × 10−197 |
| Lys_grain | QEI-sf1004407883 | LOC_Os10g08022 | Biosynthesis of amino acids | Fructose-bisphosphate aldolase isozyme | 9.1 × 10−196 |
| Lys_grain | QEI-sf0828052927 | LOC_Os08g44530 | Biosynthesis of amino acids | Dihydroxy-acid dehydratase | 2.7 × 10−301 |
| Lys_leaf | QEI-sf0103224994 | LOC_Os01g06600 | Lysine degradation | Glutaryl-CoA dehydrogenase | 2.5 × 10−152 |
| Lys_leaf | QEI-sf1016812592 | LOC_Os10g31950 | Lysine degradation | 3-ketoacyl-CoA thiolase | 7 × 10−225 |
| Lys_leaf | QEI-sf0140517811 | LOC_Os01g70170 | Biosynthesis of amino acids | 3-ketoacyl-CoA thiolase | 1.1 × 10−83 |
| Lys_leaf | QEI-sf1125165035 | LOC_Os11g42510 | Cysteine and methionine metabolism | Tyrosine aminotransferase | 1.7 × 10−166 |
| Lys_leaf | QEI-sf1220860715 | LOC_Os12g34380 | Cysteine and methionine metabolism | Glutathione synthetase | 6.2 × 10−187 |
| Lys_leaf | QEI-sf1105428802 | LOC_Os11g10140 | Tryptophan metabolism | Flavin monooxygenase | 7.5 × 10−158 |
| Lys_leaf | QEI-sf1105428802 | LOC_Os11g10170 | Tryptophan metabolism | Flavin monooxygenase | 3.5 × 10−184 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Sui, Y.; Che, Y.; Liu, L.; Liu, S.; Wang, X.; Cao, G. New Insights into the Genetic Basis of Lysine Accumulation in Rice Revealed by Multi-Model GWAS. Int. J. Mol. Sci. 2024, 25, 4667. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094667
He L, Sui Y, Che Y, Liu L, Liu S, Wang X, Cao G. New Insights into the Genetic Basis of Lysine Accumulation in Rice Revealed by Multi-Model GWAS. International Journal of Molecular Sciences. 2024; 25(9):4667. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094667
Chicago/Turabian StyleHe, Liqiang, Yao Sui, Yanru Che, Lihua Liu, Shuo Liu, Xiaobing Wang, and Guangping Cao. 2024. "New Insights into the Genetic Basis of Lysine Accumulation in Rice Revealed by Multi-Model GWAS" International Journal of Molecular Sciences 25, no. 9: 4667. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25094667