
The Integration of Transcriptome and Metabolome Analyses Provides Insights into the Determinants of the Wood Properties in Toona ciliata
 ,
,
Abstract
:1. Introduction
2. Results
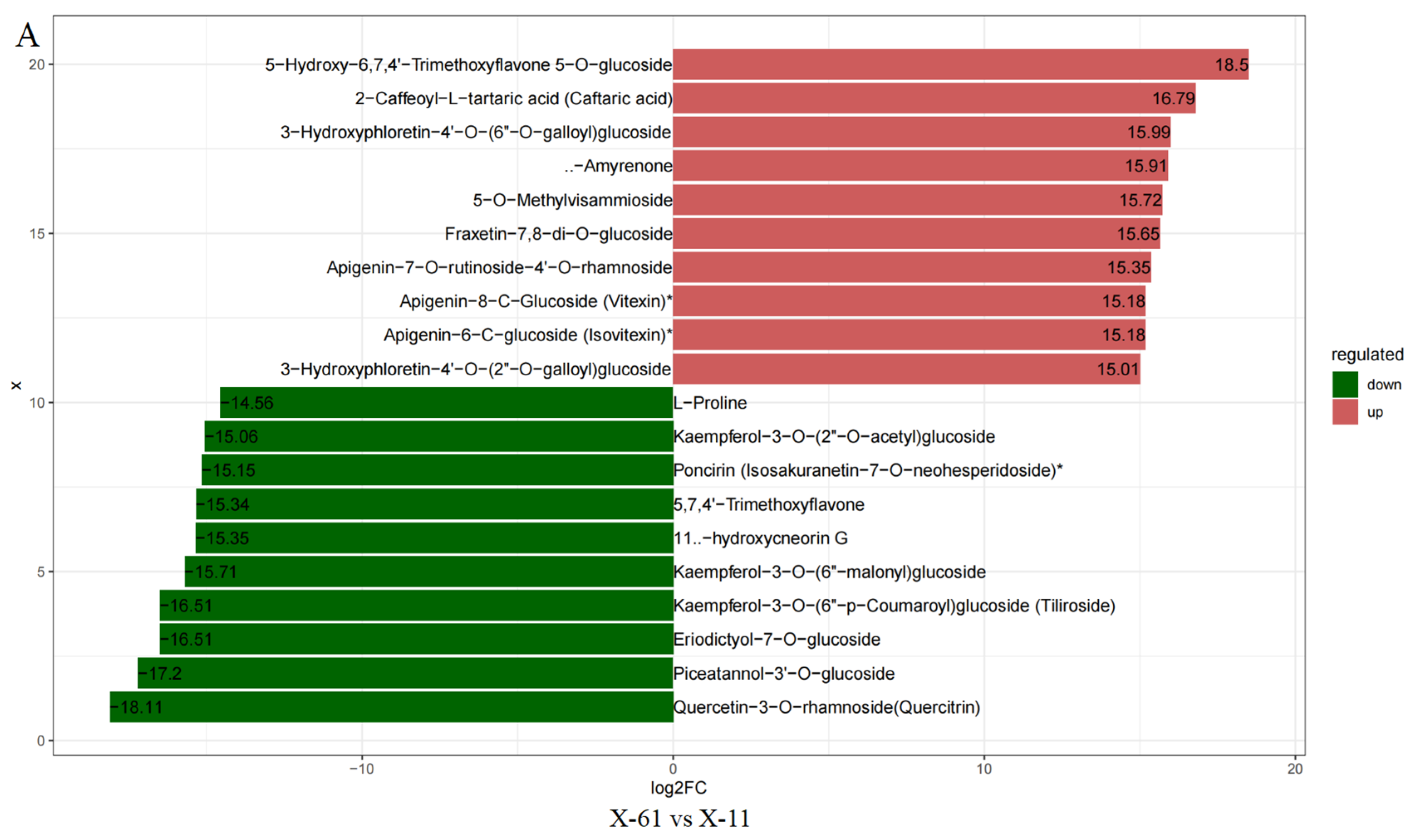
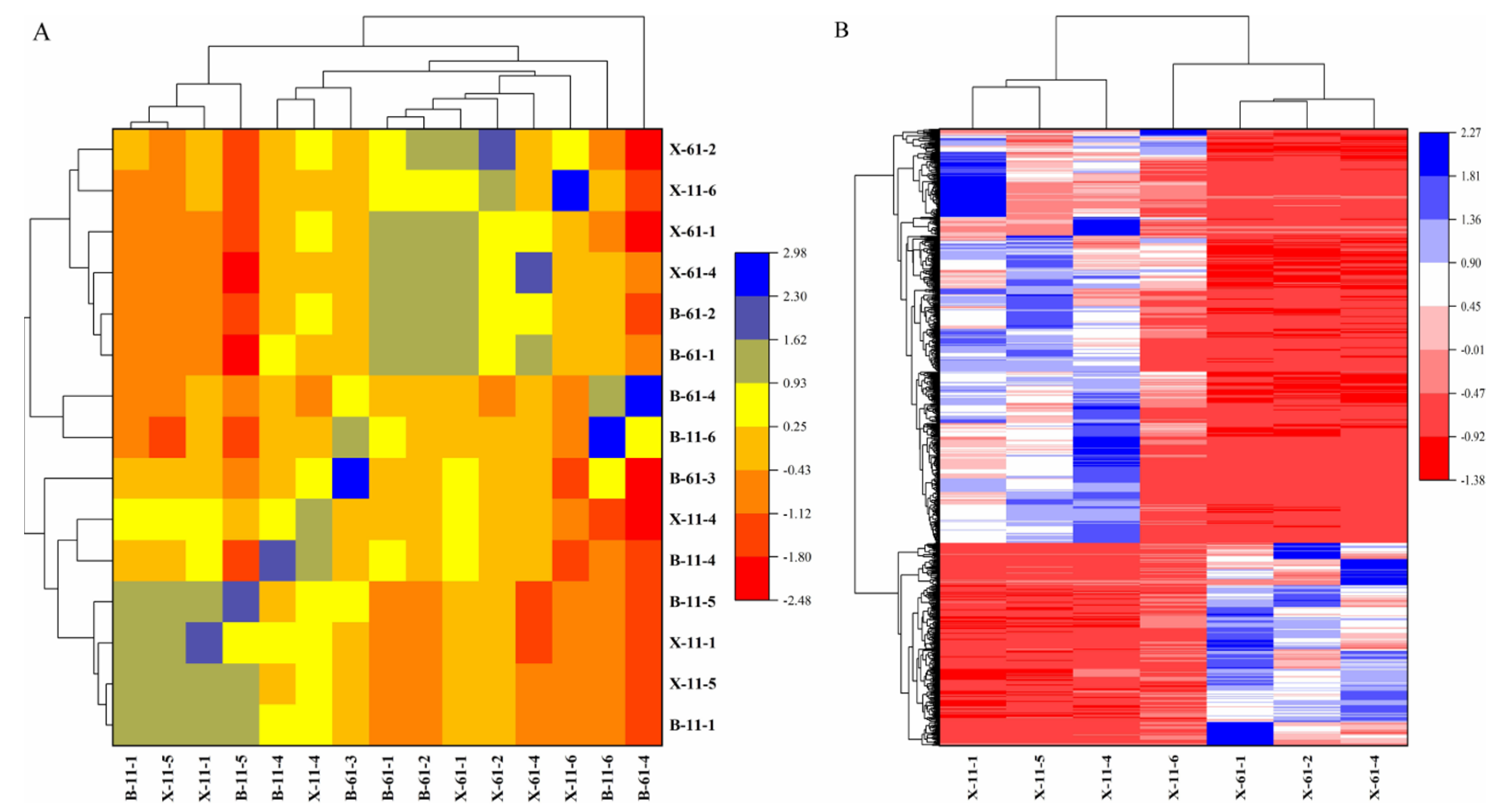
2.1. Metabolome Analysis
2.2. Transcriptome Analysis
2.2.1. Transcriptome Sequencing Results
2.2.2. Differential Gene Results
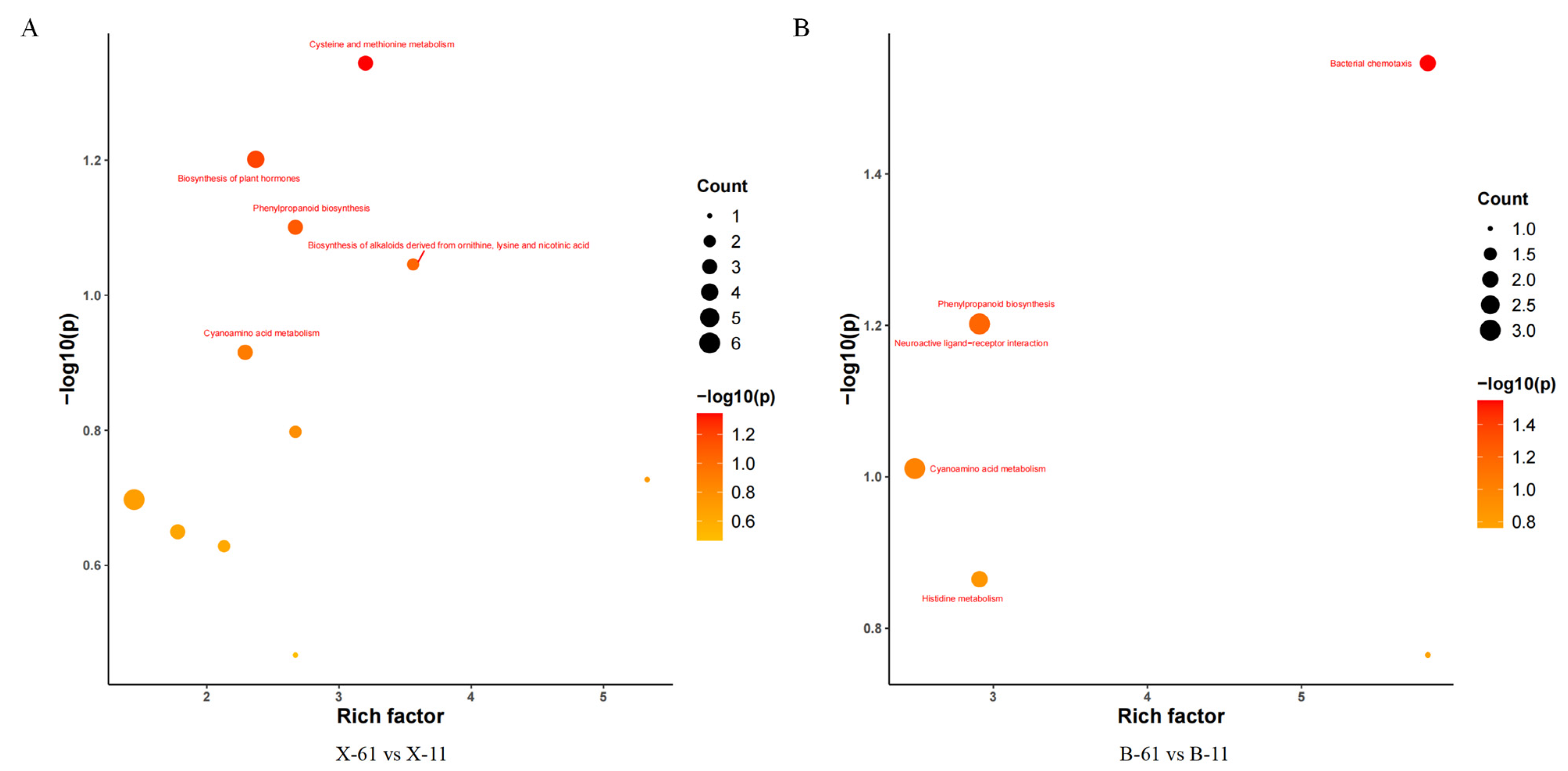
2.2.3. Differential Gene Pathway Analysis
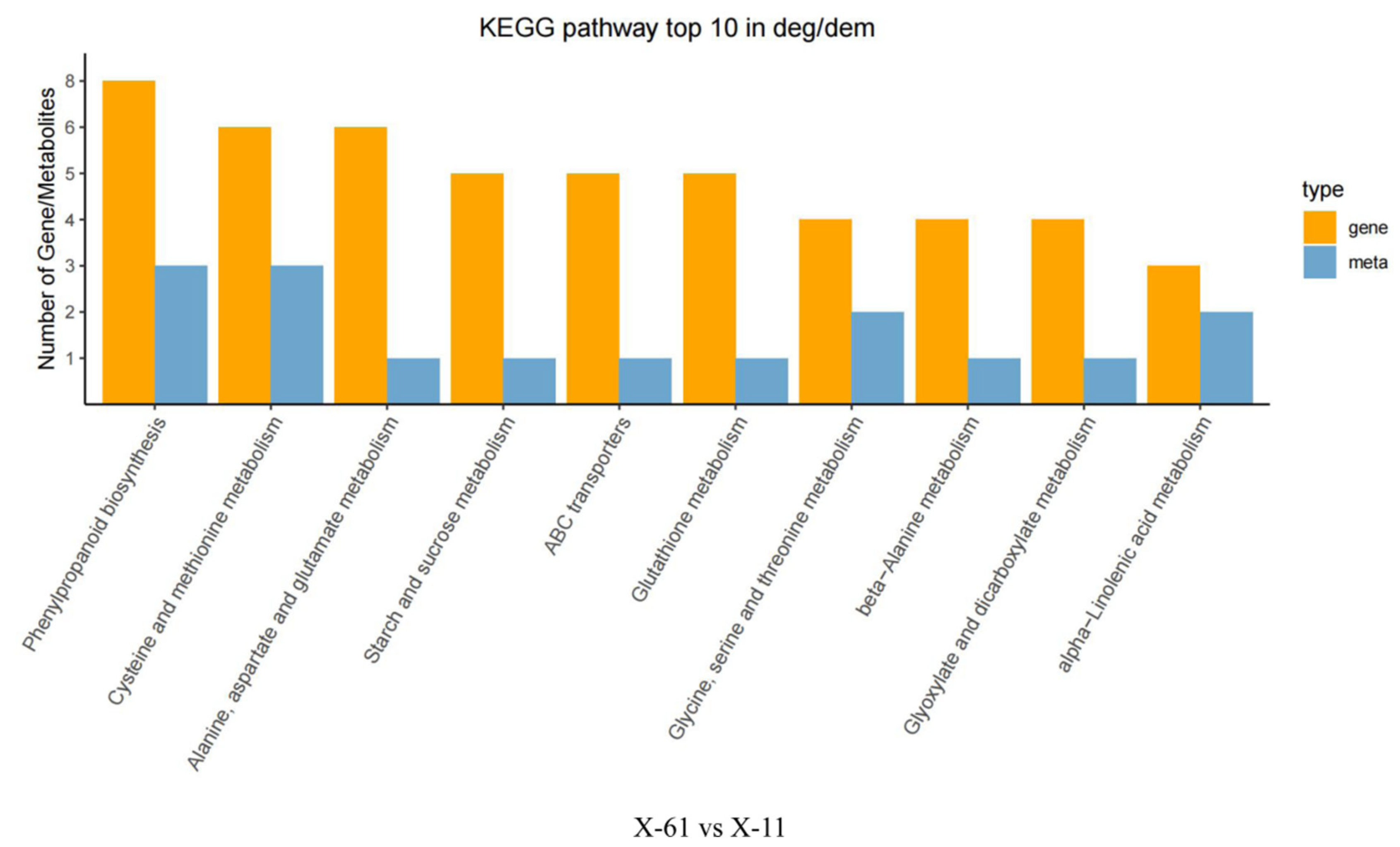
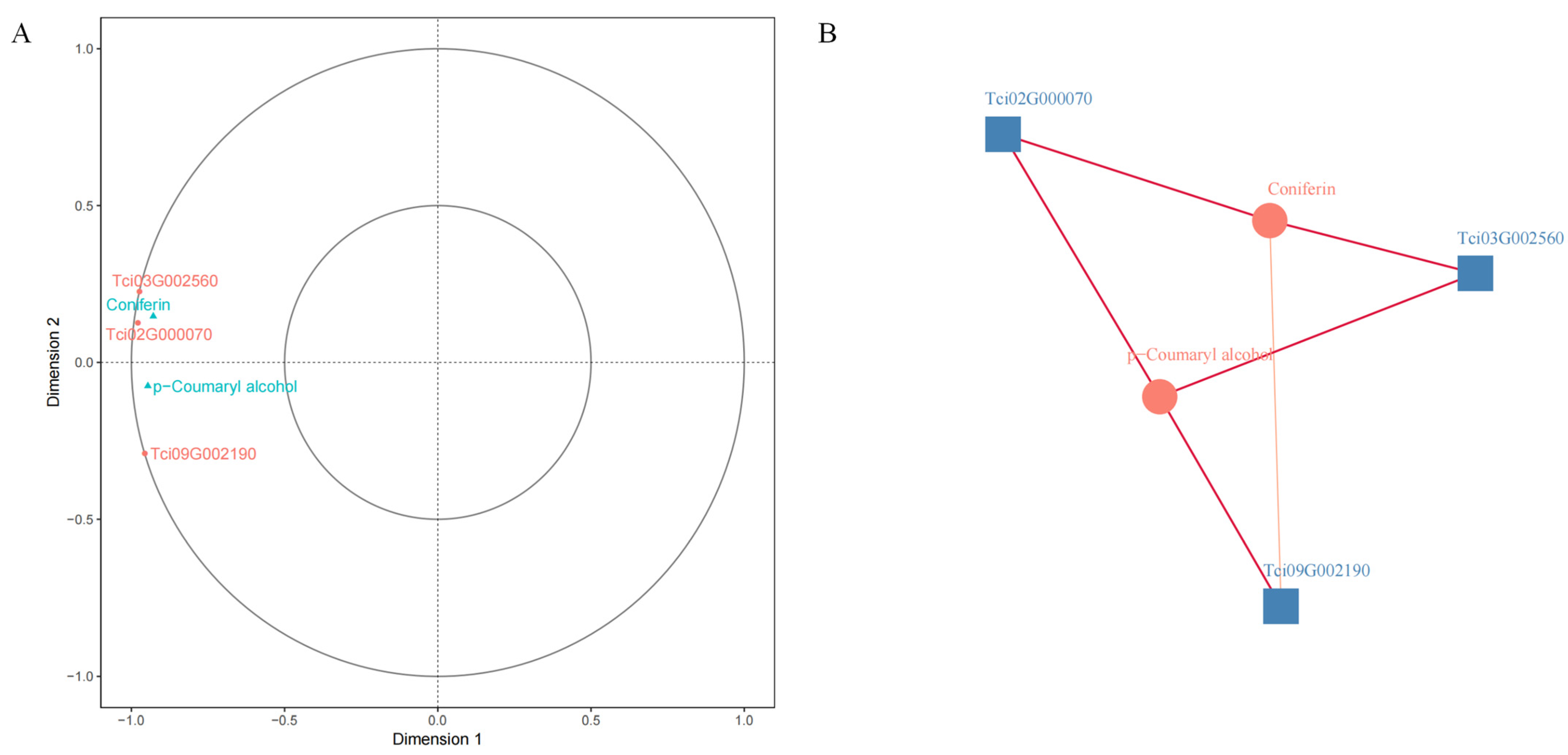
2.3. Comprehensive Analysis of the Metabolome and Transcriptome
2.4. qRT-PCR Verification
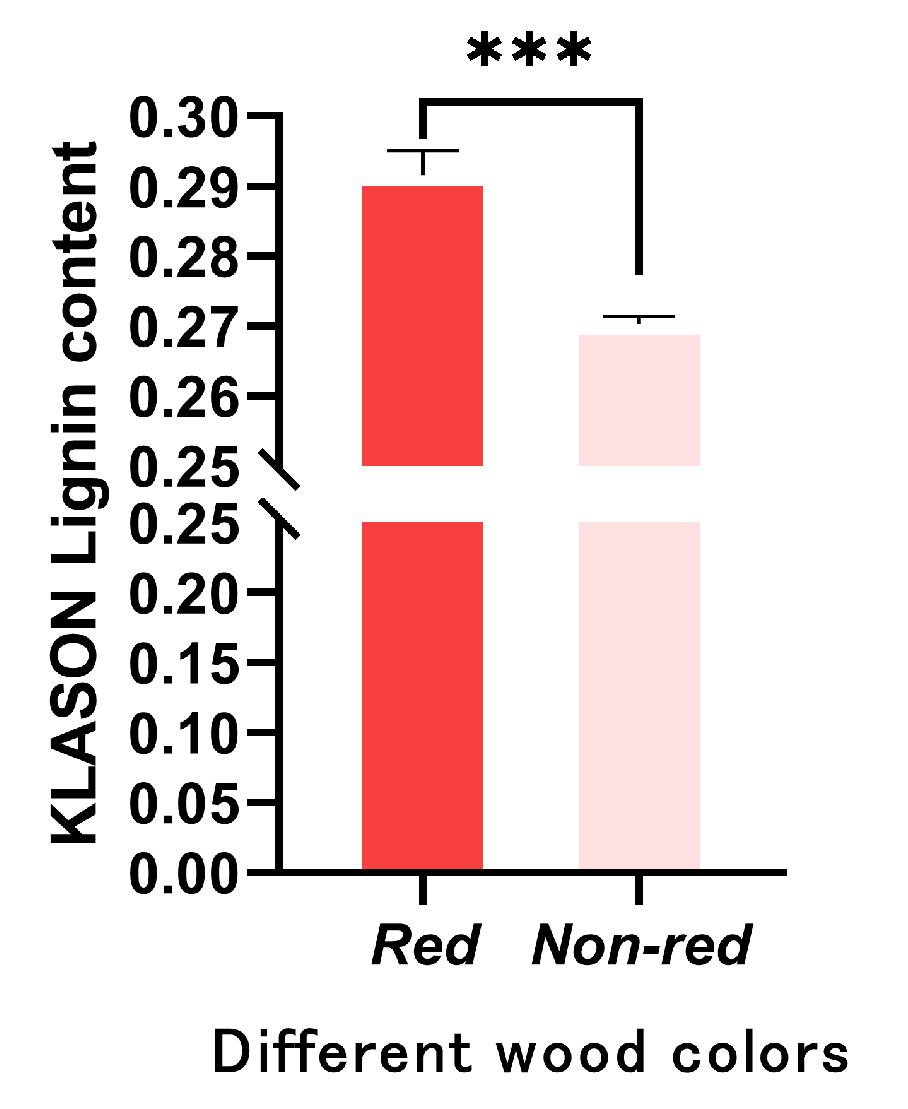
2.5. Measurement of Lignin Content
2.6. Screening of Key Genes in Lignin Synthesis
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Metabolite Determination
4.3. Transcriptome Sequencing
4.4. Transcriptome Data Analysis
4.5. Comprehensive Analysis of the Metabolome and Transcriptome
4.6. qRT-PCR Validation
4.7. Lignin Determination
4.8. Phylogenetic Analysis of the TcCADs Gene Family in T. ciliata
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, C.H. Study on Characteristics and Suitable Region Distributions for Toona ciliata and Congenerics; China Academy of Forestry Sciences: Beijing, China, 2018. [Google Scholar]
- Liu, Q.; Arnold, R.J.; Yang, S.Z.; Wu, J.Y.; Li, Z.H.; Li, Y.; Cheng, Y. Foliar application of exogenous polyamines to ameliorate drought induced oxidative damage and physiological inhibition in Toona ciliata seedlings. Aust. For. 2019, 82, 139–150. [Google Scholar] [CrossRef]
- Song, H.; Li, Y.; Wang, Z.; Duan, Z.; Wang, Y.; Yang, E.; Que, Q.; Chen, X.; Li, P. Transcriptome profiling of Toona ciliata young stems in response to Hypsipyla robusta Moore. Front. Plant Sci. 2022, 25, 950945. [Google Scholar] [CrossRef] [PubMed]
- Li, P. Study on Geographic Variation and Genetic Diversity of Toona ciliata Roem. Ph.D. Dissertation, Beijing Forestry University, Beijing, China, 2015. [Google Scholar]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Fernie, A.R. Metabolomics 20 years on: What have we learned and what hurdles remain? Plant J. 2018, 94, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef] [PubMed]
- Pandohe, J.; Kyereh, E.; Kulshrestha, S.; Xu, B.; Mahomoodally, M.F. Review of the recent developments in metabolomics-based phytochemical research. Crit. Rev. Food Sci. Nutr. 2023, 63, 3734–3749. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.J.; Zhang, M.M.; Liu, C.H.; Liu, G.; Yu, S. Progress of transcriptome sequencing of woody oil plants. Biol. Plant. 2023, 67, 189–199. [Google Scholar] [CrossRef]
- Liu, Y. Wood Color Regulation Technology of Robinia Psoudoacacia Based on Coordination Reaction. Master’s Dissertation, Beijing Forestry University, Beijing, China, 2020. [Google Scholar]
- Meints, T.; Meints, T.; Stingl, R.; Hansmann, C. Wood colour of central European wood species: CIELAB characterisation and colour intensification. Eur. Wood Wood Prod. 2017, 75, 499–509. [Google Scholar] [CrossRef]
- Louzada, J.L.P.; Fonseca, F.M. The heritability of wood density components in Pinus pinaster Ait. and the implications for tree breeding. Ann. For. Sci. 2002, 59, 867–873. [Google Scholar] [CrossRef]
- Celedon, J.M.; Bohlmann, J. An extended model of heartwood secondary metabolism informed by functional genomics. Tree Physiol. 2018, 38, 311–319. [Google Scholar] [CrossRef]
- Qiu, Q.D. Screening of Indexes for Evaluating Paulownia Wood Color and Selection of Paulownia Individuals with Excellent Wood Color. Ph.D. Thesis, China Academy of Forestry Sciences, Beijing, China, 2014. [Google Scholar]
- Faleva, A.V.; Belesov, A.V.; Kozhevnikov, A.Y.; Falev, D.I.; Chukhchin, D.G.; Novozhilov, E.V. Analysis of the functional group composition of the spruce and birch phloem lignin. Int. J. Biol. Macromol. 2021, 166, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwang, S.; Lee, S.M. Metabolic engineering for the utilization of carbohydrate portions of lignocellulosic biomass. Metab. Eng. 2022, 71, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Kacikova, D.; Kacik, F.; Cabalova, I.; Durkovic, J. Effects of thermal treatment on chemical, mechanical and colour traits in Norway spruce wood. Bioresour. Technol. 2013, 144, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.; Johansson, C.I.; Saddler, J.; Beatson, R. The nature of chromophores in high-extractives mechanical pulps: Western red cedar (Thuja plicata Donn) chemithermomechanical pulp (CTMP). Holzforschung 2006, 60, 365–371. [Google Scholar] [CrossRef]
- Jiang, X.M. Development of the Concept, Policy, Technical Problems of Valuable Broad-leaved Tree Species in Jiangxi Province and Countermeasures; Jiangxi Forestry Science and Technology: Jiangxi, China, 2013; pp. 3–8. [Google Scholar]
- Luo, W.Y.; Luo, P.; Liu, Y.J. Choice and Development of the Fine and Valuable Hardwood Tree Species in Tropical and South Subtropical Regions of China. Chin. J. Trop. Agric. 2010, 30, 15–21. [Google Scholar]
- Gall, D.L.; Ralph, J.; Donohue, T.J.; Noguera, D.R. Biochemical transformation of lignin for deriving valued commodities from lignocellulose. Curr. Opin. Biotechnol. 2017, 45, 120–126. [Google Scholar] [CrossRef]
- Mota, T.R.; de Souza, W.R.; Oliveira, D.M.; Martins, P.K.; Sampaio, B.L.; Vinecky, F.; Ribeiro, A.P.; Duarte, K.E.; Pacheco, T.F.; Monteiro, N.K.V.; et al. Suppression of a BAHD acyltransferase decreases p-coumaroyl on arabinoxylan and improves biomass digestibility in the model grass Setaria viridis. Plant J. 2021, 105, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Vanholme, R.; De Meester, B.; Ralph, J.; Boerjan, W. Lignin biosynthesis and its integration into metabolism. Curr. Opin. Biotechnol. 2019, 56, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Vanholme, R.; Cesarino, I.; Rataj, K.; Xiao, Y.; Sundin, L.; Goeminne, G.; Kim, H.; Cross, J.; Morreel, K.; Araujo, P.; et al. Caffeoyl shikimate esterase (CSE) is an enzyme in the lignin biosynthetic pathway in Arabidopsis. Science 2013, 341, 1103–1106. [Google Scholar] [CrossRef]
- Özparpucu, M.; Gierlinger, N.; Burgert, I.; Van Acker, R.; Vanholme, R.; Boerjan, W.; Pilate, G.; Dejardin, A.; Ruggeberg, M. The effect of altered lignin composition on mechanical properties of CINNAMYL ALCOHOL DEHYDROGENASE (CAD) deficient poplars. Planta 2018, 247, 887–897. [Google Scholar] [CrossRef]
- Gong, Y.; Xu, M.Q. Advance in cinnamyl alcohol dehydrogenase as a key enzyme of lignin biosynthesis. Biotechnol. Bull. 2010, 4, 47–49. [Google Scholar]
- Knight, M.E.; Halpin, C.; Schuch, W. Identification and characterisation of cDNA clones encoding cinnamyl alcohol dehydrogenase from tobacco. Plant Mol. Biol. 1992, 19, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.R.; Li, X.D.; Zhou, X.Y.; Li, X.R.; Yang, Y.; Li, B. Expression of GhCAD6 Gene in Cotton Fiber and the Effect on Structural Components. Acta Bot. Boreali-Occident. Sin. 2019, 39, 1114–1120. [Google Scholar]
- Hu, L.; Zhang, X.; Ni, H.; Yuan, F.; Zhang, S. Identification and Functional Analysis of CAD Gene Family in Pomegranate (Punica granatum). Genes 2022, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, S.F.; Siqueira, M.S.; Garon, G.C.; Alves de Lima, L.G.; de Almeida Svartman, R.M.; Cesarino, I. The lignin toolbox of the model grass Setaria viridis. Plant Mol. Biol. 2019, 101, 235–255. [Google Scholar]
- Raes, J.; Rohde, A.; Christensen, J.H.; Van de Peer, Y.; Boerjan, W. Genome-wide characterization of the lignification toolbox in Arabidopsis. Plant Physiol. 2003, 133, 1051–1071. [Google Scholar] [CrossRef] [PubMed]
- Carocha, V.; Soler, M.; Hefer, C.; Cassan-Wang, H.; Fevereiro, P.; Myburg, A.; Paiva, J.A.P.; Grima-Pettenati, J. Genome-wide analysis of the lignin toolbox of Eucalyptus grandis. New Phytol. 2015, 206, 1297–1313. [Google Scholar] [CrossRef]
- Hirano, K.; Aya, K.; Kondo, M.; Okuno, A.; Morinaka, Y.; Matsuoka, M. OsCAD2 is the major CAD gene responsible for monolignol biosynthesis in rice culm. Plant Cell Rep. 2012, 31, 91–101. [Google Scholar] [CrossRef]
- Ralph, J.; Hatfield, R.D.; Piquemal, J.; Yahiaoui, N.; Pean, M.; Lapierre, C.; Boudet, A.M. NMR characterization of altered lignins extracted from tobacco plants down-regulated for lignification enzymes cinnamylalcohol dehydrogenase and cinnamoyl-CoA reductase. Proc. Natl. Acad. Sci. USA 1998, 95, 12803–12808. [Google Scholar] [CrossRef]
- Li, L.; Cheng, X.F.; Leshkevich, J.; Umezawa, T.; Harding, S.A.; Chiang, V.L. The last step of syringyl monolignol biosynthesis in angiosperms is regulated by a novel gene encoding sinapyl alcohol dehydrogenase. Plant Cell 2001, 13, 1567–1586. [Google Scholar] [CrossRef]
- Reddy, M.S.S.; Chen, F.; Shadle, G.; Jackson, L.; Aljoe, H.; Dixon, R.A. Targeted dowm-regulaion of cytocthrome P450 enzymes for forage quality improvement in alfalfa(Medicago sativa L.). Proc. Natl. Acad. Sci. USA 2005, 102, 16573–16578. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Reddy, M.S.S.; Temple, S.; Jackson, L.; Shadle, G.; Dixon, R.A. Multi-site genetic modulation of monolignol biosynthesis suggests new routes for formation of syringyl lignin and wall-bound ferulic acid in alfalfa (Medicago sativa L.). Plant J. 2006, 48, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, J.; Chen, F.; Jackson, L.; Shadle, G.; Dixon, R. Multi-site genetic modulation of monolignol biosynthesis in alfalfa(Medicago sativa): Effects on lignin composition in specific cell types. New Phytol. 2008, 179, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q. Lignification: Flexibility, Biosynthesis and Regulation. Trends Plant Sci. 2016, 21, 713–721. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| #ID | FDR | log2FC | Regulated |
|---|---|---|---|
| Tci25G012690 | 0.002352978 | 4.569650782 | up |
| Tci04G002670 | 0.002322835 | 4.009840126 | up |
| Tci28G006780 | 5.62 × 10−7 | 3.940942152 | up |
| Tci24G013450 | 9.85 × 10−5 | 3.663677081 | up |
| Tci18G013710 | 0.000243023 | 3.472814536 | up |
| Tci17G009830 | 1.95 × 10−5 | −5.371115339 | down |
| Tci03G000040 | 4.31 × 10−12 | −4.738548588 | down |
| Tci24G007120 | 0.000123915 | −4.351597937 | down |
| Tci23G017350 | 0.006712827 | −4.19246887 | down |
| Tci03G006960 | 1.87 × 10−7 | −4.10552472 | down |
| Tci09G002190 | 5.47 × 10−5 | −4.668484043 | down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Wu, J.; Kong, W.; Zhou, Y.; Ye, C.; Yuan, Q.; Zhang, Y.; Li, P. The Integration of Transcriptome and Metabolome Analyses Provides Insights into the Determinants of the Wood Properties in Toona ciliata. Int. J. Mol. Sci. 2024, 25, 4541. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25084541
Wang Z, Wu J, Kong W, Zhou Y, Ye C, Yuan Q, Zhang Y, Li P. The Integration of Transcriptome and Metabolome Analyses Provides Insights into the Determinants of the Wood Properties in Toona ciliata. International Journal of Molecular Sciences. 2024; 25(8):4541. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25084541
Chicago/Turabian StyleWang, Zhi, Jinsong Wu, Weijia Kong, Yu Zhou, Chunyi Ye, Qianyun Yuan, Yongjia Zhang, and Pei Li. 2024. "The Integration of Transcriptome and Metabolome Analyses Provides Insights into the Determinants of the Wood Properties in Toona ciliata" International Journal of Molecular Sciences 25, no. 8: 4541. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25084541