
The Genomic Intersection of Oligodendrocyte Dynamics in Schizophrenia and Aging Unravels Novel Pathological Mechanisms and Therapeutic Potentials


Abstract
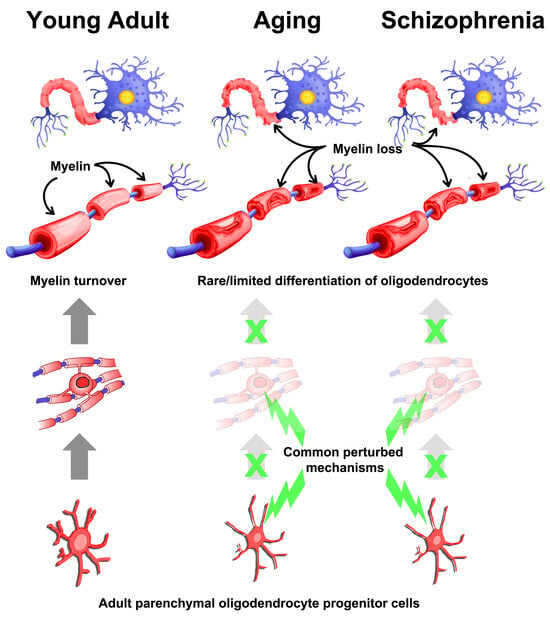
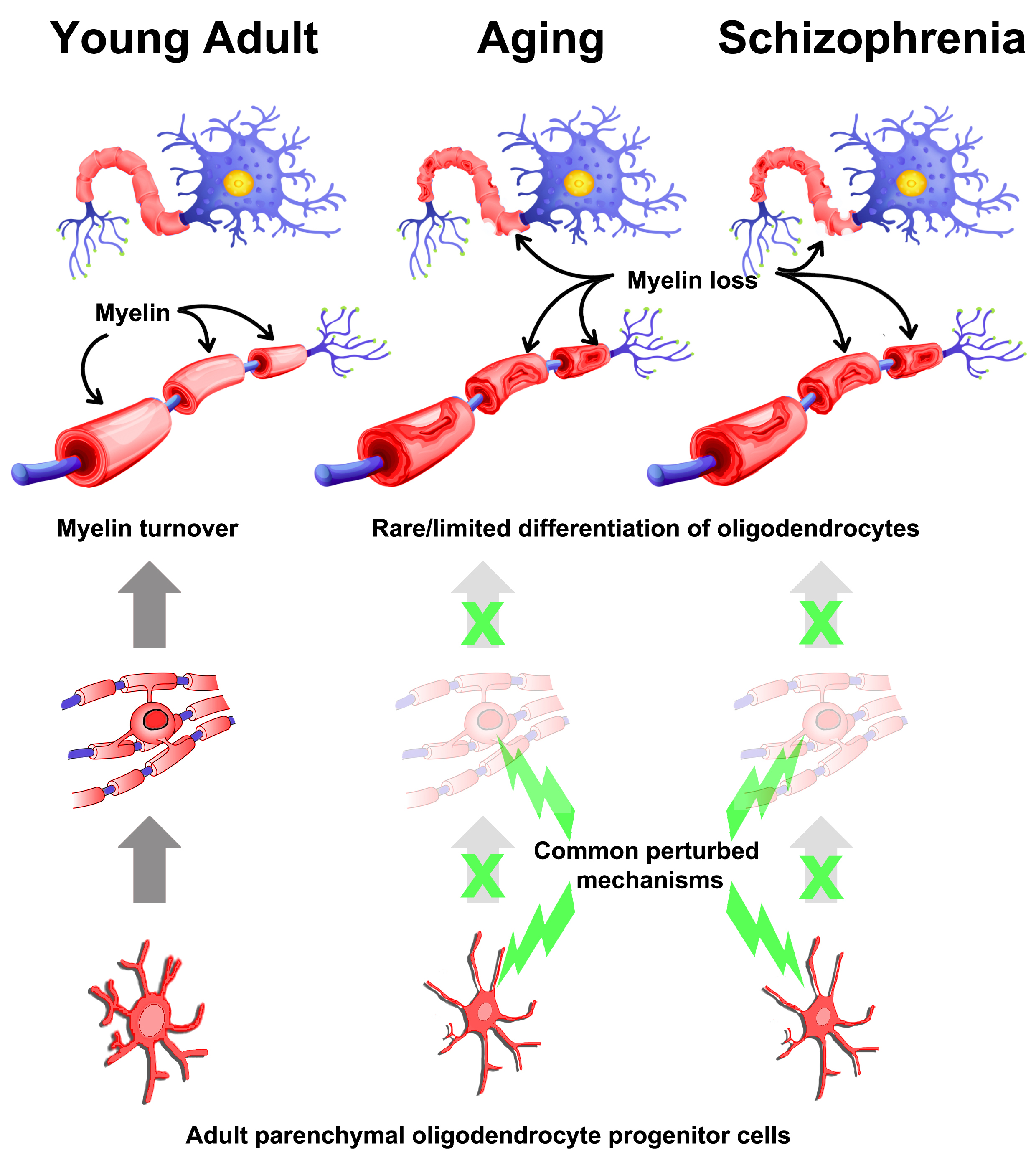
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. The Impact of Aging on OLs
1.2. Further Evidence of the Impact of Schizophrenia and Aging on White Matter from Imaging Studies
1.3. Dissecting the Relationship with Aging- and Schizophrenia-Associated Genes Using Publicly Available scRNA-Seq Datasets
2. Mechanistic Insights into OL Lineage Cells from Genome-Wide Omics: Further Bridging Aging and Schizophrenia
2.1. Insights from Upstream Receptor Signalling to Intracellular Processes in OLs in Schizophrenia Contexts
2.2. Revisiting Publicly Available Datasets for Gathering Further Insights into Schizophrenia Pathology in OLs
3. Emerging Avenues in Schizophrenia Research
3.1. Dysregulated miRNAs Expression in OLs in Schizophrenia
3.2. Altered Neuroimmune Interactions Involving Microglia and Astrocytes in Schizophrenia
3.3. Environmental Factors and Their Impact on OL Function in Schizophrenia
4. The Growing Relationship of Schizophrenia with Other Neurodegenerative Diseases: Showcasing Multiple Sclerosis
5. Looking to the Future for Schizophrenia Therapies and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laursen, T.M.; Nordentoft, M.; Mortensen, P.B. Excess Early Mortality in Schizophrenia. Annu. Rev. Clin. Psychol. 2014, 10, 425–448. [Google Scholar] [CrossRef]
- Holleran, L.; Kelly, S.; Alloza, C.; Agartz, I.; Andreassen, O.A.; Arango, C.; Banaj, N.; Calhoun, V.; Cannon, D.; Carr, V.; et al. The Relationship Between White Matter Microstructure and General Cognitive Ability in Patients With Schizophrenia and Healthy Participants in the ENIGMA Consortium. Am. J. Psychiatry 2020, 177, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Stanca, S.; Rossetti, M.; Panichi, L.B.; Bongioanni, P. The Cellular Dysfunction of the Brain–Blood Barrier from Endothelial Cells to Astrocytes: The Pathway towards Neurotransmitter Impairment in Schizophrenia. Int. J. Mol. Sci. 2024, 25, 1250. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Martin, M.; Zhou, J.-R.; Thiagalingam, S. Epigenetic Alterations of Brain Non-Neuronal Cells in Major Mental Diseases. Genes 2023, 14, 896. [Google Scholar] [CrossRef]
- Figueiredo, E.C.d.O.; Calì, C.; Petrelli, F.; Bezzi, P. Emerging evidence for astrocyte dysfunction in schizophrenia. Glia 2022, 70, 1585–1604. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Foster, A.Y.; Bujalka, H.; Emery, B. Axoglial interactions in myelin plasticity: Evaluating the relationship between neuronal activity and oligodendrocyte dynamics. Glia 2019, 67, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.S.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of Oligodendrocyte Generation and Myelination in the Human Brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Pease-Raissi, S.E.; Chan, J.R. Building a (w)rapport between neurons and oligodendroglia: Reciprocal interactions underlying adaptive myelination. Neuron 2021, 109, 1258–1273. [Google Scholar] [CrossRef]
- Tripathi, R.B.; Jackiewicz, M.; McKenzie, I.A.; Kougioumtzidou, E.; Grist, M.; Richardson, W.D. Remarkable Stability of Myelinating Oligodendrocytes in Mice. Cell Rep. 2017, 21, 316–323. [Google Scholar] [CrossRef]
- Hill, R.A.; Patel, K.D.; Goncalves, C.M.; Grutzendler, J.; Nishiyama, A. Modulation of oligodendrocyte generation during a critical temporal window after NG2 cell division. Nat. Neurosci. 2014, 17, 1518–1527. [Google Scholar] [CrossRef]
- Berkelman, R.L.; Heyward, W.L.; Stehr-Green, J.K.; Curran, J.W. Epidemiology of human immunodeficiency virus infection and acquired immunodeficiency syndrome. Am. J. Med. 1989, 86, 761–770. [Google Scholar] [CrossRef]
- Hill, R.A.; Nishiyama, A.; Hughes, E.G. Features, Fates, and Functions of Oligodendrocyte Precursor Cells. Cold Spring Harb. Perspect. Biol. 2023, 16, a041425. [Google Scholar] [CrossRef]
- Fang, L.-P.; Bai, X. Oligodendrocyte precursor cells: The multitaskers in the brain. Pflüg. Arch. Eur. J. Physiol. 2023, 475, 1035–1044. [Google Scholar] [CrossRef]
- Fekete, C.D.; Nishiyama, A. Presentation and integration of multiple signals that modulate oligodendrocyte lineage progression and myelination. Front. Cell. Neurosci. 2022, 16, 1041853. [Google Scholar] [CrossRef]
- Zou, P.; Wu, C.; Liu, T.C.-Y.; Duan, R.; Yang, L. Oligodendrocyte progenitor cells in Alzheimer’s disease: From physiology to pathology. Transl. Neurodegener. 2023, 12, 52. [Google Scholar] [CrossRef]
- Buchanan, J.; da Costa, N.M.; Cheadle, L. Emerging roles of oligodendrocyte precursor cells in neural circuit development and remodeling. Trends Neurosci. 2023, 46, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.D.; Chacon-De-La-Rocha, I.; Pieropan, F.; Papanikolau, M.; Azim, K.; Butt, A.M. Keeping the ageing brain wired: A role for purine signalling in regulating cellular metabolism in oligodendrocyte progenitors. Pflüg. Arch. Eur. J. Physiol. 2021, 473, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Czopka, T. Myelination-independent functions of oligodendrocyte precursor cells in health and disease. Nat. Neurosci. 2023, 26, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Chen, L.; Tang, T.; Qiu, M.; Xu, X. The committed oligodendrocyte precursor cell, a newly-defined intermediate progenitor cell type in oligodendroglial lineage. Glia 2023, 71, 2499–2510. [Google Scholar] [CrossRef]
- Butt, A.M.; Rivera, A.D.; Fulton, D.; Azim, K. Targeting the Subventricular Zone to Promote Myelin Repair in the Aging Brain. Cells 2022, 11, 1809. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, A.G.; Queiroz, R.M.; Ghosh, T.; McMurran, C.E.; Cubillos, J.F.; Bergles, D.E.; Fitzgerald, D.C.; Jones, C.A.; Lilley, K.S.; Glover, C.P.; et al. Changes in the Oligodendrocyte Progenitor Cell Proteome with Ageing. Mol. Cell. Proteom. 2020, 19, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.E.; Blosser, T.R.; Sullivan, Z.A.; Dulac, C.; Zhuang, X. Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 2022, 186, 194–208.e18. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.D.; Pieropan, F.; Chacon-De-La-Rocha, I.; Lecca, D.; Abbracchio, M.P.; Azim, K.; Butt, A.M. Functional genomic analyses highlight a shift in Gpr17-regulated cellular processes in oligodendrocyte progenitor cells and underlying myelin dysregulation in the aged mouse cerebrum. Aging Cell 2021, 20, e13335. [Google Scholar] [CrossRef] [PubMed]
- Cayre, M.; Falque, M.; Mercier, O.; Magalon, K.; Durbec, P. Myelin Repair: From Animal Models to Humans. Front. Cell. Neurosci. 2021, 15, 604865. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dallenga, T.; Winkler, A.; Roemer, S.; Maruschak, B.; Siebert, H.; Brück, W.; Stadelmann, C. Relationship of acute axonal damage, Wallerian degeneration, and clinical disability in multiple sclerosis. J. Neuroinflamm. 2017, 14, 57. [Google Scholar] [CrossRef]
- Chopra, S.; Shaw, M.; Shaw, T.; Sachdev, P.S.; Anstey, K.J.; Cherbuin, N. More highly myelinated white matter tracts are associated with faster processing speed in healthy adults. NeuroImage 2017, 171, 332–340. [Google Scholar] [CrossRef]
- O’sullivan, M.; Jones, D.K.; Summers, P.E.; Morris, R.G.; Williams, S.C.R.; Markus, H.S. Evidence for cortical “disconnection” as a mechanism of age-related cognitive decline. Neurology 2001, 57, 632–638. [Google Scholar] [CrossRef]
- Bartzokis, G. Age-related myelin breakdown: A developmental model of cognitive decline and Alzheimer’s disease. Neurobiol. Aging 2003, 25, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Salthouse, T.A. The processing-speed theory of adult age differences in cognition. Psychol. Rev. 1996, 103, 403–428. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ranadive, N.; Kinra, M.; Nampoothiri, M.; Arora, D.; Mudgal, J. An Overview on Chemotherapy-induced Cognitive Impairment and Potential Role of Antidepressants. Curr. Neuropharmacol. 2020, 18, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Mercier, O.; Quilichini, P.P.; Magalon, K.; Gil, F.; Ghestem, A.; Richard, F.; Boudier, T.; Cayre, M.; Durbec, P. Transient demyelination causes long-term cognitive impairment, myelin alteration and network synchrony defects. Glia 2024, 72, 960–981. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, J.; Yang, Z.; Zhang, F.; Liu, L.; Wang, P.; Biswal, B.B. Altered white matter functional pathways in Alzheimer’s disease. Cereb. Cortex 2024, 34, bhad505. [Google Scholar] [CrossRef]
- Depp, C.; Sun, T.; Sasmita, A.O.; Spieth, L.; Berghoff, S.A.; Nazarenko, T.; Overhoff, K.; Steixner-Kumar, A.A.; Subramanian, S.; Arinrad, S.; et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 2023, 618, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Wright, R. Myelin and Alzheimer’s disease. Nat. Neurosci. 2023, 26, 2048. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Long, J.; Li, W.; Wang, X.; Hu, N.; Zhao, X.; Sun, T. White matter changes in Parkinson’s disease. npj Parkinson’s Dis. 2023, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Myelin damage links brain ageing to amyloid-β deposition. Nat. Rev. Neurol. 2023, 19, 457. [Google Scholar] [CrossRef]
- Cetin-Karayumak, S.; Di Biase, M.A.; Chunga, N.; Reid, B.; Somes, N.; Lyall, A.E.; Kelly, S.; Solgun, B.; Pasternak, O.; Vangel, M.; et al. White matter abnormalities across the lifespan of schizophrenia: A harmonized multi-site diffusion MRI study. Mol. Psychiatry 2019, 25, 3208–3219. [Google Scholar] [CrossRef]
- van Erp, T.G.; Walton, E.; Hibar, D.P.; Schmaal, L.; Jiang, W.; Glahn, D.C.; Pearlson, G.D.; Yao, N.; Fukunaga, M.; Hashimoto, R.; et al. Cortical Brain Abnormalities in 4474 Individuals With Schizophrenia and 5098 Control Subjects via the Enhancing Neuro Imaging Genetics Through Meta Analysis (ENIGMA) Consortium. Biol. Psychiatry 2018, 84, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Kubicki, M.; McCarley, R.W.; Shenton, M.E. Evidence for white matter abnormalities in schizophrenia. Curr. Opin. Psychiatry 2005, 18, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Cropley, V.L.; Klauser, P.; Lenroot, R.K.; Bruggemann, J.; Sundram, S.; Bousman, C.; Pereira, A.; Di Biase, M.A.; Weickert, T.W.; Weickert, C.S.; et al. Accelerated Gray and White Matter Deterioration With Age in Schizophrenia. Am. J. Psychiatry 2017, 174, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Schnack, H.G.; van Haren, N.E.; Nieuwenhuis, M.; Pol, H.E.H.; Cahn, W.; Kahn, R.S. Accelerated Brain Aging in Schizophrenia: A Longitudinal Pattern Recognition Study. Am. J. Psychiatry 2016, 173, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, J.M.; Rogers, B.P.; Blackford, J.U.; Heckers, S.; Woodward, N.D. Accelerated Aging of Functional Brain Networks Supporting Cognitive Function in Psychotic Disorders. Biol. Psychiatry 2019, 86, 240–248. [Google Scholar] [CrossRef]
- Koshiyama, D.; Fukunaga, M.; Okada, N.; Morita, K.; Nemoto, K.; Usui, K.; Yamamori, H.; Yasuda, Y.; Fujimoto, M.; Kudo, N.; et al. White matter microstructural alterations across four major psychiatric disorders: Mega-analysis study in 2937 individuals. Mol. Psychiatry 2019, 25, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.W.; Dennis, N.A.; Buchler, N.G.; White, L.E.; Madden, D.J.; Cabeza, R. Assessing the effects of age on long white matter tracts using diffusion tensor tractography. NeuroImage 2009, 46, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Lu, P.H.; Heydari, P.; Couvrette, A.; Lee, G.J.; Kalashyan, G.; Freeman, F.; Grinstead, J.W.; Villablanca, P.; Finn, J.P.; et al. Multimodal Magnetic Resonance Imaging Assessment of White Matter Aging Trajectories Over the Lifespan of Healthy Individuals. Biol. Psychiatry 2012, 72, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Gozdas, E.; Fingerhut, H.; Chromik, L.C.; O’hara, R.; Reiss, A.L.; Hosseini, S.M.H. Focal white matter disruptions along the cingulum tract explain cognitive decline in amnestic mild cognitive impairment (aMCI). Sci. Rep. 2020, 10, 10213. [Google Scholar] [CrossRef]
- Slater, D.A.; Melie-Garcia, L.; Preisig, M.; Kherif, F.; Lutti, A.; Draganski, B. Evolution of white matter tract microstructure across the life span. Hum. Brain Mapp. 2019, 40, 2252–2268. [Google Scholar] [CrossRef]
- Brickman, A.M.; Meier, I.B.; Korgaonkar, M.S.; Provenzano, F.A.; Grieve, S.M.; Siedlecki, K.L.; Wasserman, B.T.; Williams, L.M.; Zimmerman, M.E. Testing the white matter retrogenesis hypothesis of cognitive aging. Neurobiol. Aging 2012, 33, 1699–1715. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.R.; Ritchie, S.J.; Tucker-Drob, E.M.; Liewald, D.C.; Hagenaars, S.P.; Davies, G.; Wardlaw, J.M.; Gale, C.R.; Bastin, M.E.; Deary, I.J. Ageing and brain white matter structure in 3,513 UK Biobank participants. Nat. Commun. 2016, 7, 13629. [Google Scholar] [CrossRef] [PubMed]
- Kochunov, P.; Ganjgahi, H.; Winkler, A.; Kelly, S.; Shukla, D.K.; Du, X.; Jahanshad, N.; Rowland, L.; Sampath, H.; Patel, B.; et al. Heterochronicity of white matter development and aging explains regional patient control differences in schizophrenia. Hum. Brain Mapp. 2016, 37, 4673–4688. [Google Scholar] [CrossRef]
- Rivera, A.; Azim, K.; Butt, A. Resolving the age-related decline in central nervous system myelin turnover and drug discovery for oligodendroglial rejuvenation. Neural Regen. Res. 2022, 17, 2677–2678. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.M.; Nicaise, A.M.; Bongarzone, E.R.; Givogri, M.; Reiter, C.R.; Heintz, O.; Jellison, E.R.; Sutter, P.A.; TeHennepe, G.; Ananda, G.; et al. Astrocyte Support for Oligodendrocyte Differentiation can be Conveyed via Extracellular Vesicles but Diminishes with Age. Sci. Rep. 2020, 10, 828. [Google Scholar] [CrossRef]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell 2019, 25, 473–485.e8. [Google Scholar] [CrossRef] [PubMed]
- Sexton, C.E.; Walhovd, K.B.; Storsve, A.B.; Tamnes, C.K.; Westlye, L.T.; Johansen-Berg, H.; Fjell, A.M. Accelerated Changes in White Matter Microstructure during Aging: A Longitudinal Diffusion Tensor Imaging Study. J. Neurosci. 2014, 34, 15425–15436. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Tatsch, L.; Vollhardt, A.; Schneider-Axmann, T.; Raabe, F.J.; Roell, L.; Heinsen, H.; Hof, P.R.; Falkai, P.; Schmitz, C. Decreased Oligodendrocyte Number in Hippocampal Subfield CA4 in Schizophrenia: A Replication Study. Cells 2022, 11, 3242. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.F.; Ding, J.; Langston, R.G.; Shah, S.I.; Nalls, M.A.; Scholz, S.W.; Whitaker, D.T.; Auluck, P.K.; Marenco, S.; Gibbs, J.R.; et al. Divergent patterns of healthy aging across human brain regions at single-cell resolution reveal links to neurodegenerative disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Wang, Q.; Antone, J.; Alsop, E.; Reiman, R.; Funk, C.; Bendl, J.; Dudley, J.T.; Liang, W.S.; Karr, T.L.; Roussos, P.; et al. A public resource of single cell transcriptomes and multiscale networks from persons with and without Alzheimer’s disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fitzgerald, E.; Arcego, D.M.; Shen, M.J.; O’Toole, N.; Wen, X.; Nagy, C.; Mostafavi, S.; Craig, K.; Silveira, P.P.; Rayan, N.A.; et al. Sex and cell-specific gene expression in corticolimbic brain regions associated with psychiatric disorders revealed by bulk and single-nuclei RNA sequencing. eBioMedicine 2023, 95, 104749. [Google Scholar] [CrossRef] [PubMed]
- Chehimi, S.N.; Crist, R.C.; Reiner, B.C. Unraveling Psychiatric Disorders through Neural Single-Cell Transcriptomics Approaches. Genes 2023, 14, 771. [Google Scholar] [CrossRef] [PubMed]
- Ling, E.; Nemesh, J.; Goldman, M.; Kamitaki, N.; Reed, N.; Handsaker, R.E.; Genovese, G.; Vogelgsang, J.S.; Gerges, S.; Kashin, S.; et al. A concerted neuron–astrocyte program declines in ageing and schizophrenia. Nature 2024, 627, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Hamed, A.A.; Kunz, D.J.; El-Hamamy, I.; Trinh, Q.M.; Subedar, O.D.; Richards, L.M.; Foltz, W.; Bullivant, G.; Ware, M.; Vladoiu, M.C.; et al. A brain precursor atlas reveals the acquisition of developmental-like states in adult cerebral tumours. Nat. Commun. 2022, 13, 4178. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2023, 42, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Cebrian-Silla, A.; Nascimento, M.A.; Redmond, S.A.; Mansky, B.; Wu, D.; Obernier, K.; Rodriguez, R.R.; Gonzalez-Granero, S.; García-Verdugo, J.M.; Lim, D.A.; et al. Single-cell analysis of the ventricular-subventricular zone reveals signatures of dorsal and ventral adult neurogenesis. eLife 2021, 10, e67436. [Google Scholar] [CrossRef] [PubMed]
- Marcy, G.; Foucault, L.; Babina, E.; Capeliez, T.; Texeraud, E.; Zweifel, S.; Heinrich, C.; Hernandez-Vargas, H.; Parras, C.; Jabaudon, D.; et al. Single-cell analysis of the postnatal dorsal V-SVZ reveals a role for Bmpr1a signaling in silencing pallial germinal activity. Sci. Adv. 2023, 9, eabq7553. [Google Scholar] [CrossRef]
- Khandker, L.; Jeffries, M.A.; Chang, Y.-J.; Mather, M.L.; Evangelou, A.V.; Bourne, J.N.; Tafreshi, A.K.; Ornelas, I.M.; Bozdagi-Gunal, O.; Macklin, W.B.; et al. Cholesterol biosynthesis defines oligodendrocyte precursor heterogeneity between brain and spinal cord. Cell Rep. 2022, 38, 110423. [Google Scholar] [CrossRef]
- Liu, L.; Kim, S.; Buckley, M.T.; Reyes, J.M.; Kang, J.; Tian, L.; Wang, M.; Lieu, A.; Mao, M.; Rodriguez-Mateo, C.; et al. Exercise reprograms the inflammatory landscape of multiple stem cell compartments during mammalian aging. Cell Stem Cell 2023, 30, 689–705.e4. [Google Scholar] [CrossRef] [PubMed]
- Luecken, M.D.; Büttner, M.; Chaichoompu, K.; Danese, A.; Interlandi, M.; Mueller, M.F.; Strobl, D.C.; Zappia, L.; Dugas, M.; Colomé-Tatché, M.; et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 2021, 19, 41–50. [Google Scholar] [CrossRef]
- Katsel, P.; Fam, P.; Tan, W.; Khan, S.; Yang, C.; Jouroukhin, Y.; Rudchenko, S.; Pletnikov, M.V.; Haroutunian, V. Overexpression of Truncated Human DISC1 Induces Appearance of Hindbrain Oligodendroglia in the Forebrain During Development. Schizophr. Bull. 2017, 44, 515–524. [Google Scholar] [CrossRef] [PubMed]
- De Vrij, F.M.; Bouwkamp, C.G.; Gunhanlar, N.; Shpak, G.; Lendemeijer, B.; Baghdadi, M.; Gopalakrishna, S.; Ghazvini, M.; Li, T.M.; Quadri, M.; et al. Candidate CSPG4 mutations and induced pluripotent stem cell modeling implicate oligodendrocyte progenitor cell dysfunction in familial schizophrenia. Mol. Psychiatry 2018, 24, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Yalcinbas, E.A.; Ajanaku, B.; Nelson, E.D.; Garcia-Flores, R.; Montgomery, K.D.; Stolz, J.M.; Wu, J.; Divecha, H.R.; Chandra, A.; Bharadwaj, R.A.; et al. Transcriptomic analysis of the human habenula in schizophrenia. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kim, S.; Webster, M.J. Correlation analysis between genome-wide expression profiles and cytoarchitectural abnormalities in the prefrontal cortex of psychiatric disorders. Mol. Psychiatry 2008, 15, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Hakak, Y.; Walker, J.R.; Li, C.; Wong, W.H.; Davis, K.L.; Buxbaum, J.D.; Haroutunian, V.; Fienberg, A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef]
- Hagihara, H.; Ohira, K.; Takao, K.; Miyakawa, T. Transcriptomic evidence for immaturity of the prefrontal cortex in patients with schizophrenia. Mol. Brain 2014, 7, 41. [Google Scholar] [CrossRef]
- Ma, C.; Gu, C.; Huo, Y.; Li, X.; Luo, X.-J. The integrated landscape of causal genes and pathways in schizophrenia. Transl. Psychiatry 2018, 8, 67. [Google Scholar] [CrossRef]
- Pimenova, A.A.; Raj, T.; Goate, A.M. Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 2017, 83, 300–310. [Google Scholar] [CrossRef]
- Aston, C.; Jiang, L.; Sokolov, B.P. Transcriptional profiling reveals evidence for signaling and oligodendroglial abnormalities in the temporal cortex from patients with major depressive disorder. Mol. Psychiatry 2004, 10, 309–322. [Google Scholar] [CrossRef]
- Le-Niculescu, H.; Balaraman, Y.; Patel, S.; Tan, J.; Sidhu, K.; Jerome, R.; Edenberg, H.; Kuczenski, R.; Geyer, M.; Nurnberger, J.; et al. Towards understanding the schizophrenia code: An expanded convergent functional genomics approach. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2007, 144B, 129–158. [Google Scholar] [CrossRef]
- Ishimoto, T.; Ninomiya, K.; Inoue, R.; Koike, M.; Uchiyama, Y.; Mori, H. Mice lacking BCAS1, a novel myelin-associated protein, display hypomyelination, schizophrenia-like abnormal behaviors, and upregulation of inflammatory genes in the brain. Glia 2017, 65, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Mauney, S.A.; Pietersen, C.Y.; Sonntag, K.-C.; Woo, T.-U.W. Differentiation of oligodendrocyte precursors is impaired in the prefrontal cortex in schizophrenia. Schizophr. Res. 2015, 169, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhu, Z.; Ransom, B.R.; Tong, X. Oligodendrocyte lineage cells and depression. Mol. Psychiatry 2020, 26, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Gouvêa-Junqueira, D.; Falvella, A.C.B.; Antunes, A.S.L.M.; Seabra, G.; Brandão-Teles, C.; Martins-De-Souza, D.; Crunfli, F. Novel Treatment Strategies Targeting Myelin and Oligodendrocyte Dysfunction in Schizophrenia. Front. Psychiatry 2020, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Raabe, F.J.; Slapakova, L.; Rossner, M.J.; Cantuti-Castelvetri, L.; Simons, M.; Falkai, P.G.; Schmitt, A. Oligodendrocytes as A New Therapeutic Target in Schizophrenia: From Histopathological Findings to Neuron-Oligodendrocyte Interaction. Cells 2019, 8, 1496. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Su, Y.; Guo, C.; Yi, C.; Yu, B.; Chen, H.; Cui, Y.; Wang, X.; Wang, Y.; Chen, X.; et al. Pathological oligodendrocyte precursor cells revealed in human schizophrenic brains and trigger schizophrenia-like behaviors and synaptic defects in genetic animal model. Mol. Psychiatry 2022, 27, 5154–5166. [Google Scholar] [CrossRef]
- Candido, K.; Soufi, H.; Bandyopadhyay, M.; Dasgupta, S. Therapeutic Impact of Sphingosine 1-phosphate Receptor Signaling in Multiple Sclerosis. Mini-Rev. Med. Chem. 2016, 15, 1–8. [Google Scholar] [CrossRef]
- Esaki, K.; Balan, S.; Iwayama, Y.; Shimamoto-Mitsuyama, C.; Hirabayashi, Y.; Dean, B.; Yoshikawa, T. Evidence for Altered Metabolism of Sphingosine-1-Phosphate in the Corpus Callosum of Patients with Schizophrenia. Schizophr. Bull. 2020, 46, 1172–1181. [Google Scholar] [CrossRef]
- Monin, A.; Baumann, P.S.; Griffa, A.; Xin, L.; Mekle, R.; Fournier, M.; Butticaz, C.; Klaey, M.; Cabungcal, J.H.; Steullet, P.; et al. Glutathione deficit impairs myelin maturation: Relevance for white matter integrity in schizophrenia patients. Mol. Psychiatry 2014, 20, 827–838. [Google Scholar] [CrossRef]
- Rivera, A.D.; Azim, K.; Macchi, V.; Porzionato, A.; Butt, A.M.; De Caro, R. Epidermal Growth Factor Pathway in the Age-Related Decline of Oligodendrocyte Regeneration. Front. Cell. Neurosci. 2022, 16, 838007. [Google Scholar] [CrossRef]
- Hu, X.; Xiao, G.; He, L.; Niu, X.; Li, H.; Lou, T.; Hu, Q.; Yang, Y.; Xu, Q.; Wei, Z.; et al. Sustained ErbB Activation Causes Demyelination and Hypomyelination by Driving Necroptosis of Mature Oligodendrocytes and Apoptosis of Oligodendrocyte Precursor Cells. J. Neurosci. 2021, 41, 9872–9890. [Google Scholar] [CrossRef] [PubMed]
- Galvez-Contreras, A.Y.; Quiñones-Hinojosa, A.; Gonzalez-Perez, O. The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Front. Cell. Neurosci. 2013, 7, 258. [Google Scholar] [CrossRef]
- Roy, K.; Murtie, J.C.; El-Khodor, B.F.; Edgar, N.; Sardi, S.P.; Hooks, B.M.; Benoit-Marand, M.; Chen, C.; Moore, H.; O’Donnell, P.; et al. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 8131–8136. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Kamiya, A.; Yokota, Y.; Prikulis, I.; Kano, S.-I.; Hayashi-Takagi, A.; Stanco, A.; Eom, T.-Y.; Rao, S.; Ishizuka, K.; et al. Disrupted-in-Schizophrenia-1 expression is regulated by β-site amyloid precursor protein cleaving enzyme-1–neuregulin cascade. Proc. Natl. Acad. Sci. USA 2010, 107, 5622–5627. [Google Scholar] [CrossRef]
- Katsel, P.; Tan, W.; Abazyan, B.; Davis, K.L.; Ross, C.; Pletnikov, M.V.; Haroutunian, V. Expression of mutant human DISC1 in mice supports abnormalities in differentiation of oligodendrocytes. Schizophr. Res. 2011, 130, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Piñero, J.; Queralt-Rosinach, N.; Bravo, A.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, Y.-G.; Luo, X.-J. SZDB: A Database for Schizophrenia Genetic Research. Schizophr. Bull. 2016, 43, 459–471. [Google Scholar] [CrossRef]
- Kerns, D.; Vong, G.S.; Barley, K.; Dracheva, S.; Katsel, P.; Casaccia, P.; Haroutunian, V.; Byne, W. Gene expression abnormalities and oligodendrocyte deficits in the internal capsule in schizophrenia. Schizophr. Res. 2010, 120, 150–158. [Google Scholar] [CrossRef]
- Katsel, P.; Davis, K.L.; Haroutunian, V. Variations in myelin and oligodendrocyte-related gene expression across multiple brain regions in schizophrenia: A gene ontology study. Schizophr. Res. 2005, 79, 157–173. [Google Scholar] [CrossRef]
- McCullumsmith, R.E.; Gupta, D.; Beneyto, M.; Kreger, E.; Haroutunian, V.; Davis, K.L.; Meadorwoodruff, J. Expression of transcripts for myelination-related genes in the anterior cingulate cortex in schizophrenia. Schizophr. Res. 2007, 90, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.; Davis, K.L.; Li, C.; Tan, W.; Greenstein, E.; Hoffman, L.B.K.; Haroutunian, V. Abnormal Indices of Cell Cycle Activity in Schizophrenia and their Potential Association with Oligodendrocytes. Neuropsychopharmacology 2008, 33, 2993–3009. [Google Scholar] [CrossRef]
- Pillinger, T.; Beck, K.; Gobjila, C.; Donocik, J.G.; Jauhar, S.; Howes, O.D. Impaired Glucose Homeostasis in First-Episode Schizophrenia. JAMA Psychiatry 2017, 74, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.E.; Banner, J.; Jensen, S.E. Cardiovascular disease in patients with severe mental illness. Nat. Rev. Cardiol. 2020, 18, 136–145. [Google Scholar] [CrossRef] [PubMed]
- DE Hert, M.; Schreurs, V.; Vancampfort, D.; VAN Winkel, R. Metabolic syndrome in people with schizophrenia: A review. World Psychiatry 2009, 8, 15–22. [Google Scholar] [CrossRef]
- Soreq, L.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; Patani, R.; Ule, J. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef]
- Azim, K.; Rivera, A.; Raineteau, O.; Butt, A.M. GSK3β regulates oligodendrogenesis in the dorsal microdomain of the subventricular zone via Wnt-β-catenin signaling. Glia 2014, 62, 778–789. [Google Scholar] [CrossRef]
- Azim, K.; Fischer, B.; Hurtado-Chong, A.; Draganova, K.; Cantù, C.; Zemke, M.; Sommer, L.; Butt, A.; Raineteau, O. Persistent Wnt/β-Catenin Signaling Determines Dorsalization of the Postnatal Subventricular Zone and Neural Stem Cell Specification into Oligodendrocytes and Glutamatergic Neurons. Stem Cells 2014, 32, 1301–1312. [Google Scholar] [CrossRef]
- Azim, K.; Butt, A.M. GSK3β negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 2011, 59, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Hoseth, E.Z.; Krull, F.; Dieset, I.; Mørch, R.H.; Hope, S.; Gardsjord, E.S.; Steen, N.E.; Melle, I.; Brattbakk, H.-R.; Steen, V.M.; et al. Exploring the Wnt signaling pathway in schizophrenia and bipolar disorder. Transl. Psychiatry 2018, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.N.; Rizavi, H.S.; Tripathi, M.; Ren, X. Region-specific dysregulation of glycogen synthase kinase-3β and β-catenin in the postmortem brains of subjects with bipolar disorder and schizophrenia. Bipolar Disord. 2014, 17, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Seidensticker, M.J.; Behrens, J. Biochemical interactions in the wnt pathway. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2000, 1495, 168–182. [Google Scholar] [CrossRef]
- Azim, K.; Angonin, D.; Marcy, G.; Pieropan, F.; Rivera, A.; Donega, V.; Cantù, C.; Williams, G.; Berninger, B.; Butt, A.M.; et al. Pharmacogenomic identification of small molecules for lineage specific manipulation of subventricular zone germinal activity. PLOS Biol. 2017, 15, e2000698. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. Lithium: A potential therapeutic strategy in obsessive–compulsive disorder by targeting the canonical WNT/β pathway. Transl. Psychiatry 2021, 11, 204. [Google Scholar] [CrossRef]
- Snitow, M.E.; Bhansali, R.S.; Klein, P.S. Lithium and Therapeutic Targeting of GSK-3. Cells 2021, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Damri, O.; Shemesh, N.; Agam, G. Is There Justification to Treat Neurodegenerative Disorders by Repurposing Drugs? The Case of Alzheimer’s Disease, Lithium, and Autophagy. Int. J. Mol. Sci. 2020, 22, 189. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Draghi, N.A.; Resh, M.D. Signaling from Integrins to Fyn to Rho Family GTPases Regulates Morphologic Differentiation of Oligodendrocytes. J. Neurosci. 2004, 24, 7140–7149. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.W.; Kim, J.Y.; Yoon, S.O. Activation of Rac GTPase by p75 Is Necessary for c-junN-Terminal Kinase-Mediated Apoptosis. J. Neurosci. 2002, 22, 156–166. [Google Scholar] [CrossRef]
- Parkinson, D.B.; Bhaskaran, A.; Arthur-Farraj, P.; Noon, L.A.; Woodhoo, A.; Lloyd, A.C.; Feltri, M.L.; Wrabetz, L.; Behrens, A.; Mirsky, R.; et al. c-Jun is a negative regulator of myelination. J. Cell Biol. 2008, 181, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Hu, C.; Yang, W.; Guo, D.; Li, C.; Shen, W.; Liu, X.; Aijun, H.; Dan, W.; He, C. NMDA receptor couples Rac1-GEF Tiam1 to direct oligodendrocyte precursor cell migration. Glia 2013, 61, 2078–2099. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tu, X.; Joeng, K.S.; Hilton, M.J.; Williams, D.A.; Long, F. Rac1 Activation Controls Nuclear Localization of β-catenin during Canonical Wnt Signaling. Cell 2008, 133, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Takagi, A.; Araki, Y.; Nakamura, M.; Vollrath, B.; Duron, S.G.; Yan, Z.; Kasai, H.; Huganir, R.L.; Campbell, D.A.; Sawa, A. PAKs inhibitors ameliorate schizophrenia-associated dendritic spine deterioration in vitro and in vivo during late adolescence. Proc. Natl. Acad. Sci. USA 2014, 111, 6461–6466. [Google Scholar] [CrossRef]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 2010, 13, 327–332. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/20139976%5Cnhttp://www.nature.com/neuro/journal/v13/n3/pdf/nn.2487.pdf (accessed on 10 January 2022). [CrossRef] [PubMed]
- Thurnherr, T.; Benninger, Y.; Wu, X.; Chrostek, A.; Krause, S.M.; Nave, K.-A.; Franklin, R.J.M.; Brakebusch, C.; Suter, U.; Relvas, J.B. Cdc42 and Rac1 Signaling Are Both Required for and Act Synergistically in the Correct Formation of Myelin Sheaths in the CNS. J. Neurosci. 2006, 26, 10110–10119. [Google Scholar] [CrossRef] [PubMed]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef]
- Ngo, C.; Kothary, R. MicroRNAs in oligodendrocyte development and remyelination. J. Neurochem. 2022, 162, 310–321. [Google Scholar] [CrossRef]
- Santarelli, D.M.; Carroll, A.P.; Cairns, H.M.; Tooney, P.A.; Cairns, M.J. Schizophrenia-associated MicroRNA–Gene Interactions in the Dorsolateral Prefrontal Cortex. Genom. Proteom. Bioinform. 2019, 17, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Lee, S.-Y.; Scarr, E.; Yu, Y.-H.; Lin, Y.-T.; Hwang, T.-J.; Hsieh, M.H.; Liu, C.-C.; Chien, Y.-L.; Udawela, M.; et al. Aberrant expression of microRNAs as biomarker for schizophrenia: From acute state to partial remission, and from peripheral blood to cortical tissue. Transl. Psychiatry 2016, 6, e717. [Google Scholar] [CrossRef]
- He, X.; Yu, Y.; Awatramani, R.; Lu, Q.R. Unwrapping Myelination by MicroRNAs. Neuroscientist 2011, 18, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Crowley, J.J. A comprehensive review of the genetic and biological evidence supports a role for MicroRNA-137 in the etiology of schizophrenia. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2017, 177, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-H.; Du, Y.; Chen, L.; Cheng, Y. Glial Cell Abnormalities in Major Psychiatric Diseases: A Systematic Review of Postmortem Brain Studies. Mol. Neurobiol. 2022, 59, 1665–1692. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chung, W.-S. Astrocyte-dependent circuit remodeling by synapse phagocytosis. Curr. Opin. Neurobiol. 2023, 81, 102732. [Google Scholar] [CrossRef] [PubMed]
- Haroutunian, V.; Katsel, P.; Roussos, P.; Davis, K.L.; Altshuler, L.L.; Bartzokis, G. Myelination, oligodendrocytes, and serious mental illness. Glia 2014, 62, 1856–1877. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.-G.; Steiner, J.; Guest, P.C.; Dobrowolny, H.; Bogerts, B. Glial cells as key players in schizophrenia pathology: Recent insights and concepts of therapy. Schizophr. Res. 2015, 161, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Radulescu, E.; Jaffe, A.E.; Straub, R.E.; Chen, Q.; Shin, J.H.; Hyde, T.M.; Kleinman, J.E.; Weinberger, D.R. Identification and prioritization of gene sets associated with schizophrenia risk by co-expression network analysis in human brain. Mol. Psychiatry 2018, 25, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.V.; Svistunov, A.A.; Chubarev, V.N.; Sologova, S.S.; Mukhortova, P.; Levushkin, D.; Somasundaram, S.G.; Kirkland, C.E.; Bachurin, S.O.; Aliev, G. Alterations of Astrocytes in the Context of Schizophrenic Dementia. Front. Pharmacol. 2020, 10, 1612. [Google Scholar] [CrossRef] [PubMed]
- McEwan, F.; Glazier, J.D.; Hager, R. The impact of maternal immune activation on embryonic brain development. Front. Neurosci. 2023, 17, 1146710. [Google Scholar] [CrossRef]
- Guardiola-Ripoll, M.; Fatjó-Vilas, M. A Systematic Review of the Human Accelerated Regions in Schizophrenia and Related Disorders: Where the Evolutionary and Neurodevelopmental Hypotheses Converge. Int. J. Mol. Sci. 2023, 24, 3597. [Google Scholar] [CrossRef]
- Barichello, T.; Simoes, L.R.; Quevedo, J.; Zhang, X.Y. Microglial Activation and Psychotic Disorders: Evidence from Pre-clinical and Clinical Studies. Curr. Top. Behav. Neurosci. 2020, 44, 161–205. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Chen, T.; Yan, A.; Xiao, J.; Xie, Y.-L.; Yuan, J.; Chen, P.; Wong, A.O.-L.; Zhang, Y.; Wong, N.-K. Poly(I:C) Challenge Alters Brain Expression of Oligodendroglia-Related Genes of Adult Progeny in a Mouse Model of Maternal Immune Activation. Front. Mol. Neurosci. 2020, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Crombie, G.K.; Palliser, H.K.; Shaw, J.C.; Hanley, B.A.; Moloney, R.A.; Hirst, J.J. Prenatal Stress Induces Translational Disruption Associated with Myelination Deficits. Dev. Neurosci. 2023, 45, 290–308. [Google Scholar] [CrossRef] [PubMed]
- Masroor, A.; Khorochkov, A.; Prieto, J.; Singh, K.B.; Nnadozie, M.C.; Abdal, M.; Shrestha, N.; Abe, R.A.M.; Mohammed, L. Unraveling the Association Between Schizophrenia and Substance Use Disorder-Predictors, Mechanisms and Treatment Modifications: A Systematic Review. Cureus 2021, 13, e16722. [Google Scholar] [CrossRef] [PubMed]
- Zamberletti, E.; Beggiato, S.; Steardo, L.; Prini, P.; Antonelli, T.; Ferraro, L.; Rubino, T.; Parolaro, D. Alterations of prefrontal cortex GABAergic transmission in the complex psychotic-like phenotype induced by adolescent delta-9-tetrahydrocannabinol exposure in rats. Neurobiol. Dis. 2014, 63, 35–47. [Google Scholar] [CrossRef]
- Yang, H.-J.; Wang, L.; Cheng, Q.; Xu, H. Abnormal Behaviors and Microstructural Changes in White Matter of Juvenile Mice Repeatedly Exposed to Amphetamine. Schizophr. Res. Treat. 2011, 2011, 542896. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Agrawal, K.; Hui, H.; Qin, Y.; Rana, T.M. Glial cell diversity and methamphetamine-induced neuroinflammation in human cerebral organoids. Mol. Psychiatry 2020, 26, 1194–1207. [Google Scholar] [CrossRef]
- Marrie, R.A.; Fisk, J.D.; Yu, B.N.; Leung, S.; Elliott, L.; Caetano, P.; Warren, S.; Evans, C.; Wolfson, C.; Svenson, L.W.; et al. Mental comorbidity and multiple sclerosis: Validating administrative data to support population-based surveillance. BMC Neurol. 2013, 13, 16. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Harbo, H.F.; Wang, Y.; Thompson, W.K.; Schork, A.J.; Mattingsdal, M.; Zuber, V.; Bettella, F.; Ripke, S.; Kelsoe, J.R.; et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: Differential involvement of immune-related gene loci. Mol. Psychiatry 2014, 20, 207–214. [Google Scholar] [CrossRef]
- Pouget, J.G.; Han, B.; Wu, Y.; Mignot, E.; Ollila, H.M.; Barker, J.; Spain, S.; Dand, N.; Trembath, R.; Martin, J.; et al. Cross-disorder analysis of schizophrenia and 19 immune-mediated diseases identifies shared genetic risk. Hum. Mol. Genet. 2019, 28, 3498–3513. [Google Scholar] [CrossRef]
- Benros, M.E.; Pedersen, M.G.; Rasmussen, H.; Eaton, W.W.; Nordentoft, M.; Mortensen, P.B. A Nationwide Study on the Risk of Autoimmune Diseases in Individuals With a Personal or a Family History of Schizophrenia and Related Psychosis. Am. J. Psychiatry 2014, 171, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Ahangari, M.; Everest, E.; Nguyen, T.-H.; Verrelli, B.C.; Webb, B.T.; Bacanu, S.-A.; Turanli, E.T.; Riley, B.P. Genome-wide analysis of schizophrenia and multiple sclerosis identifies shared genomic loci with mixed direction of effects. Brain Behav. Immun. 2022, 104, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Misiak, B.; Samochowiec, J.; Kowalski, K.; Gaebel, W.; Bassetti, C.L.A.; Chan, A.; Gorwood, P.; Papiol, S.; Dom, G.; Volpe, U.; et al. The future of diagnosis in clinical neurosciences: Comparing multiple sclerosis and schizophrenia. Eur. Psychiatry 2023, 66, 1–25. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. Serum-Based Biomarkers in Neurodegeneration and Multiple Sclerosis. Biomedicines 2022, 10, 1077. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, I.; McCall, M.N. Systematic exploration of cell morphological phenotypes associated with a transcriptomic query. Nucleic Acids Res. 2018, 46, e116. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Watanabe, E.; Fukuchi, M. Psychiatric Neural Networks and Precision Therapeutics by Machine Learning. Biomedicines 2021, 9, 403. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Knol, M.J.; Knol, M.J.; Wang, R.; Wang, R.; Mishra, A.; Mishra, A.; Liu, D.; Liu, D.; et al. Epigenetic and integrative cross-omics analyses of cerebral white matter hyperintensities on MRI. Brain 2022, 146, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.H.; Wang, C.; Soh, W.K.; Rajapakse, J.C. Combining Neuroimaging and Omics Datasets for Disease Classification Using Graph Neural Networks. Front. Neurosci. 2022, 16, 866666. [Google Scholar] [CrossRef]
- Jain, P.R.; Yates, M.; de Celis, C.R.; Drineas, P.; Jahanshad, N.; Thompson, P.; Paschou, P. Multiomic approach and Mendelian randomization analysis identify causal associations between blood biomarkers and subcortical brain structure volumes. NeuroImage 2023, 284, 120466. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera, A.D.; Normanton, J.R.; Butt, A.M.; Azim, K. The Genomic Intersection of Oligodendrocyte Dynamics in Schizophrenia and Aging Unravels Novel Pathological Mechanisms and Therapeutic Potentials. Int. J. Mol. Sci. 2024, 25, 4452. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25084452
Rivera AD, Normanton JR, Butt AM, Azim K. The Genomic Intersection of Oligodendrocyte Dynamics in Schizophrenia and Aging Unravels Novel Pathological Mechanisms and Therapeutic Potentials. International Journal of Molecular Sciences. 2024; 25(8):4452. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25084452
Chicago/Turabian StyleRivera, Andrea D., John R. Normanton, Arthur M. Butt, and Kasum Azim. 2024. "The Genomic Intersection of Oligodendrocyte Dynamics in Schizophrenia and Aging Unravels Novel Pathological Mechanisms and Therapeutic Potentials" International Journal of Molecular Sciences 25, no. 8: 4452. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25084452