
Palladium-Catalyzed Esterification of Aryl Fluorosulfates with Aryl Formates

 ,
,
Abstract
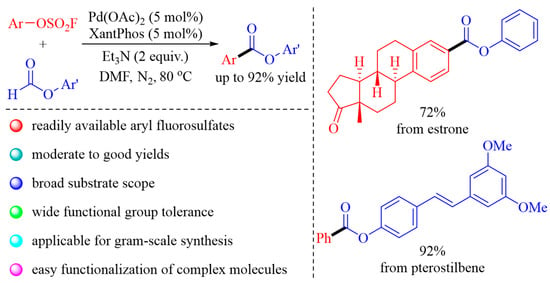
:
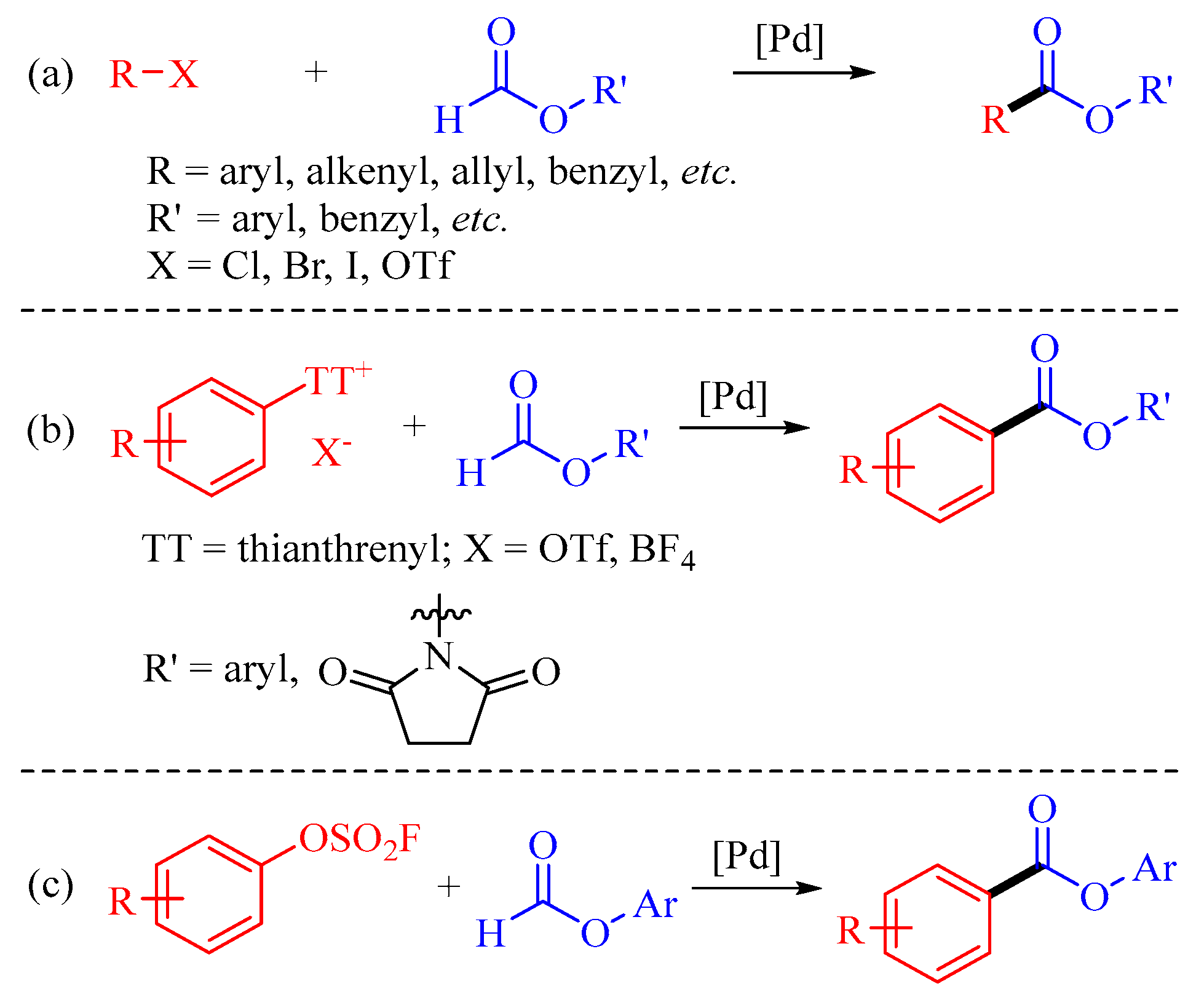
1. Introduction
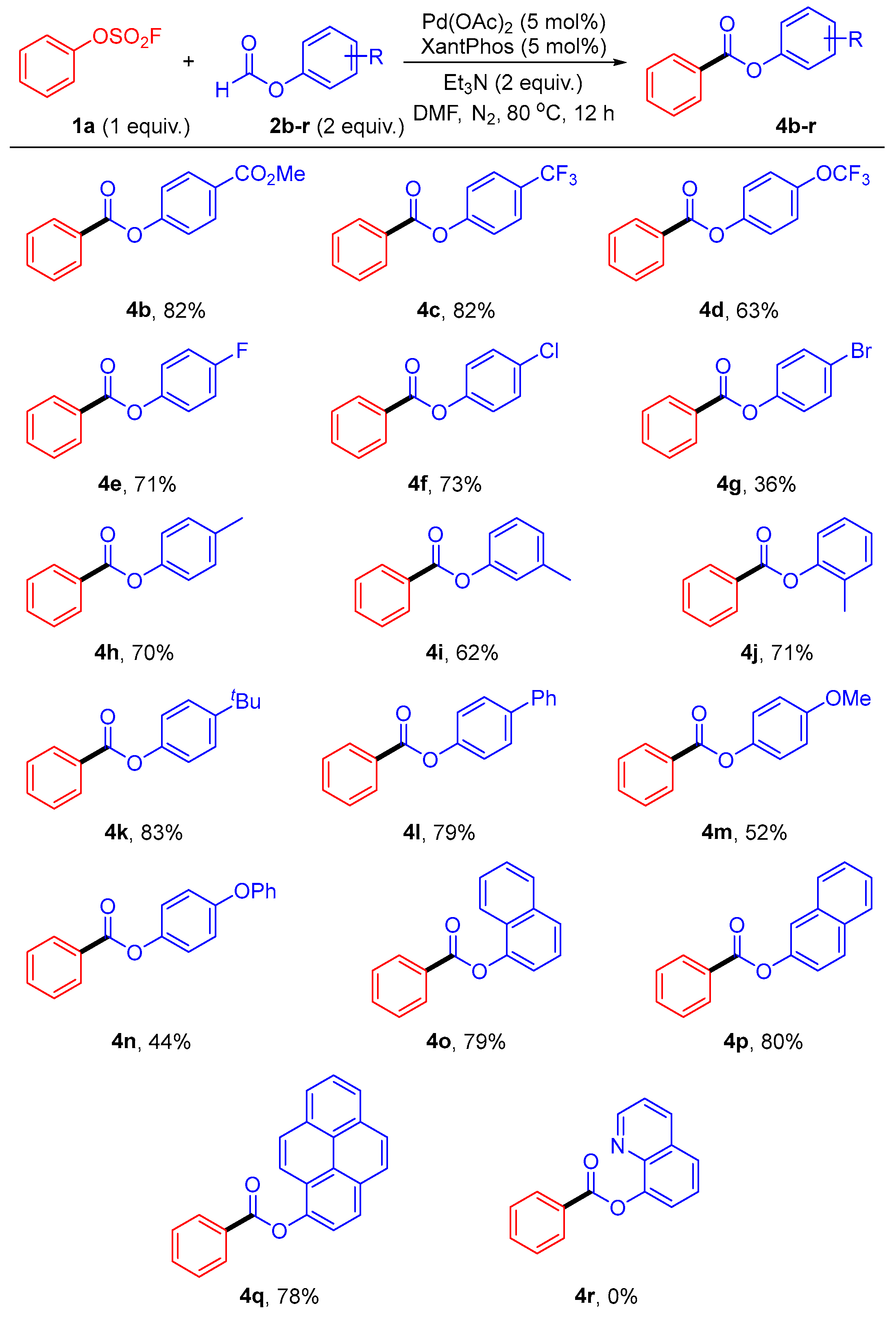
2. Results
3. Materials and Methods
3.1. General Information
3.2. Experimental
3.2.1. General Procedure for the Synthesis of Aryl Fluorosulfates 1a–r
3.2.2. General Procedure for the Synthesis of Aryl Formates 2a–s
3.2.3. General Procedure for the Cross-Coupling of Aryl Fluorosulfates with Aryl Formates
3.2.4. Gram-Scale Synthesis of Aryl Fluorosulfate 1a with Aryl Formate 2a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Otera, J. Esterification: Methods, Reactions, and Applications; John Wiley & Sons: Chichester, UK, 2003. [Google Scholar]
- Gooßen, L.J.; Rodríguez, N.; Gooßen, K. Carboxylic Acids as Substrates in Homogeneous Catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar] [CrossRef] [PubMed]
- Majji, G.; Rout, S.K.; Rajamanickam, S.; Guin, S.; Patel, B.K. Synthesis of Esters via sp3 C–H Functionalization. Org. Biomol. Chem. 2016, 14, 8178–8211. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Zheng, F. Synthesis of Carboxylic Acids and Esters from CO2. Top. Curr. Chem. 2017, 375, 4. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylative Coupling Reactions between Ar–X and Carbon Nucleophiles. Chem. Soc. Rev. 2011, 40, 4986–5009. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.B.; Geng, H.Q.; Wu, X.F. The Chemistry of CO: Carbonylation. Chem 2019, 5, 526–552. [Google Scholar] [CrossRef]
- Zhu, C.; Liu, J.; Li, M.B.; Bäckvall, J.E. Palladium-Catalyzed Oxidative Dehydrogenative Carbonylation Reactions Using Carbon Monoxide and Mechanistic Overviews. Chem. Soc. Rev. 2020, 49, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Brennführer, A.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylation Reactions of Aryl Halides and Related Compounds. Angew. Chem. Int. Ed. 2009, 48, 4114–4133. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Neumann, H.; Beller, M. Synthesis of Heterocycles via Palladium-Catalyzed Carbonylations. Chem. Rev. 2013, 113, 1–35. [Google Scholar] [CrossRef]
- Wu, X.F.; Neumann, H.; Beller, M. Palladium-Catalyzed Oxidative Carbonylation Reactions. ChemSusChem 2013, 6, 229–241. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, H.; Lei, A. Oxidative Carbonylation Reactions: Organometallic Compounds (R–M) or Hydrocarbons (R–H) as Nucleophiles. Angew. Chem. Int. Ed. 2011, 50, 10788–10799. [Google Scholar] [CrossRef]
- Barnard, C.F.J. Palladium-Catalyzed Carbonylation–A Reaction Come of Age. Organometallics 2008, 27, 5402–5422. [Google Scholar] [CrossRef]
- Konishi, H.; Manabe, K. Recent Progress on Catalytic Heck Carbonylations Using Carbon Monoxide Surrogates. Tetrahedron Lett. 2019, 60, 151147. [Google Scholar] [CrossRef]
- Morimoto, T.; Kakiuchi, K. Evolution of Carbonylation Catalysis: No Need for Carbon Monoxide. Angew. Chem. Int. Ed. 2004, 43, 5580–5588. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, L.C.; Wu, X.F. Carbonylative Synthesis of Heterocycles Involving Diverse CO Surrogates. Chem. Commun. 2020, 56, 6016–6030. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Konishi, H.; Manabe, K. Palladium-Catalyzed Carbonylation of Aryl, Alkenyl, and Allyl Halides with Phenyl Formate. Org. Lett. 2012, 14, 3100–3103. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Konishi, H.; Manabe, K. Trichlorophenyl Formate: Highly Reactive and Easily Accessible Crystalline CO Surrogate for Palladium-Catalyzed Carbonylation of Aryl/Alkenyl Halides and Triflates. Org. Lett. 2012, 14, 5370–5373. [Google Scholar] [CrossRef]
- Fujihara, T.; Hosoki, T.; Katafuchi, Y.; Iwai, T.; Terao, J.; Tsuji, Y. Palladium-Catalyzed Esterification of Aryl Halides Using Aryl Formates without the Use of External Carbon Monoxide. Chem. Commun. 2012, 48, 8012–8014. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Leng, Y.; Yang, F.; Wang, S.; Wu, Y. Palladacycle-Catalyzed Carbonylation of Aryl Iodides or Bromides with Aryl Formates. Chin. J. Chem. 2013, 31, 1488–1494. [Google Scholar] [CrossRef]
- Barre, A.; Tintas, M.L.; Alix, F.; Gembus, V.; Papamicael, C.; Levacher, V. Palladium-Catalyzed Carbonylation of (Hetero)Aryl, Alkenyl and Allyl Halides by Means of N-Hydroxysuccinimidyl Formate as CO Surrogate. J. Org. Chem. 2015, 80, 6537–6544. [Google Scholar] [CrossRef]
- Konishi, H.; Hoshino, F.; Manabe, K. Practical Synthesis of Axially Chiral Dicarboxylates via Pd-Catalyzed External-CO-Free Carbonylation. Chem. Pharm. Bull. 2016, 64, 1438–1441. [Google Scholar] [CrossRef]
- Konishi, H.; Matsubara, M.; Mori, K.; Tokiwa, T.; Arulmozhiraja, S.; Yamamoto, Y.; Ishikawa, Y.; Hashimoto, H.; Shigeta, Y.; Tokiwa, H.; et al. Mechanistic Insight into Weak Base-Catalyzed Generation of Carbon Monoxide from Phenyl Formate and Its Application to Catalytic Carbonylation at Room Temperature without Use of External Carbon Monoxide Gas. Adv. Synth. Catal. 2017, 359, 3592–3601. [Google Scholar] [CrossRef]
- Lai, M.; Qi, X.; Wu, X.F. Palladium-Catalyzed Carbonylative Synthesis of Benzyl Benzoates Employing Benzyl Formates as Both CO Surrogates and Benzyl Alcohol Sources. Eur. J. Org. Chem. 2019, 23, 3776–3777. [Google Scholar] [CrossRef]
- Maddocks, C.J.; Aathimanikandan, S.V.; Richardson, J.; Ruble, J.C. Quinolin-8-yl Formate: A New Option for Small-Scale Carbonylation Reactions in Microwave Reactors. Synlett 2020, 31, 1608–1612. [Google Scholar]
- Shen, Z.; Huo, Y.W.; Qi, X.; Wu, X.F. Palladium-Catalyzed Thiocarbonylative Synthesis of α,β-Unsaturated Thioesters Using S-Aryl Thioformates as the Thioester Sources. Org. Lett. 2023, 25, 4140–4144. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Manabe, K. Formic Acid Derivatives as Practical Carbon Monoxide Surrogates for Metal-Catalyzed Carbonylation Reactions. Synlett 2014, 25, 1971–1986. [Google Scholar] [CrossRef]
- Fujihara, T.; Tsuji, Y.J. Transition-Metal Catalyzed Synthesis of Carbonyl Compounds Using Formates as Carbonyl Sources. Jpn. Petrol. Inst. 2018, 61, 1–9. [Google Scholar] [CrossRef]
- Sawant, D.N.; Wagh, Y.S.; Bhatte, K.D.; Bhanage, B.M. Palladium-Catalyzed Carbon-Monoxide-Free Aminocarbonylation of Aryl Halides Using N-Substituted Formamides as an Amide Source. J. Org. Chem. 2011, 76, 5489–5494. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.L.; Chen, Z.B.; Zhu, Y.M.; Ji, S.J. Palladium-Catalyzed Carbonylative Annulation Reactions Using Aryl Formate as a CO Source: Synthesis of 2-Substituted Indene-1,3(2H)-dione Derivatives. J. Org. Chem. 2015, 80, 10643–10650. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lan, J.; Guo, S.; You, J. Pd-Catalyzed C-H Carbonylation of (Hetero)Arenes with Formates and Intramolecular Dehydrogenative Coupling: A Shortcut to Indolo[3,2-c]coumarins. Org. Lett. 2014, 16, 5862–5865. [Google Scholar] [CrossRef]
- Chavan, S.P.; Bhanage, B.M. Carbonylative Synthesis of Phthalimides and Benzoxazinones by Using Phenyl Formate as a Carbon Monoxide Source. Eur. J. Org. Chem. 2015, 2015, 2405–2410. [Google Scholar] [CrossRef]
- Konishi, H.; Nagasea, H.; Manabe, K. Concise Synthesis of Cyclic Carbonyl Compounds from Haloarenes using Phenyl Formate as the Carbonyl Source. Chem. Commun. 2015, 51, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Chen, Z.B.; Zhang, F.L.; Zhu, Y.M. Palladium-Catalyzed Carbonylative Synthesis of Isocoumarins and Phthalides by using Phenyl Formate as a Carbon Monoxide Source. Org. Biomol. Chem. 2017, 15, 1628–1635. [Google Scholar] [CrossRef]
- Konishi, H.; Futamata, S.; Wang, X.; Manabe, K. Rapid Formation of Fluoren-9-ones via Palladium-Catalyzed External Carbon Monoxide-Free Carbonylation. Adv. Synth. Catal. 2018, 360, 1805–1809. [Google Scholar] [CrossRef]
- Cheng, C.; Wan, B.; Zhou, B.; Gu, Y.; Zhang, Y. Enantioselective Synthesis of Quaternary 3,4-Dihydroisoquinolinones via Heck Carbonylation Reactions: Development and Application to the Synthesis of Minalrestat Analogues. Chem. Sci. 2019, 10, 9853–9858. [Google Scholar] [CrossRef] [PubMed]
- Song, K.L.; Wu, B.; Gan, W.E.; Yang, W.C.; Chen, X.B.; Cao, J.; Xu, L.W. Palladium-Catalyzed Gaseous CO-Free Carbonylative C–C Bond Activation of Cyclobutanones. Org. Chem. Front. 2021, 8, 3398–3403. [Google Scholar] [CrossRef]
- Ye, H.; Wu, L.; Zhang, M.; Jiang, G.; Dai, H.; Wu, X.X. Palladium-Catalyzed Heck Cyclization/Carbonylation with Formates: Synthesis of Azaindoline-3-Acetates and Furoazaindolines. Chem. Commun. 2022, 58, 6825–6828. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Neumann, H.; Beller, M.; Wu, X.F. Aryl Formate as Bifunctional Reagent: Applications in Palladium-Catalyzed Carbonylative Coupling Reactions Using in Situ Generated CO. Angew. Chem. Int. Ed. 2014, 53, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lai, M.; Zhu, M.J.; Peng, J.B.; Ying, J.; Wu, X.F. 1-Arylvinyl Formats: A New CO Source and Ketone Source in Carbonylative Synthesis of Chalcone Derivatives. ChemCatChem 2019, 11, 5252–5255. [Google Scholar] [CrossRef]
- Xiang, X.B.; Wang, S.; Xu, T.; Chen, S. Palladium(II)-Catalyzed Regioselective Hydroesterification of 1,3-Conjugated Enynes with Aryl Formates. Org. Lett. 2023, 25, 587–591. [Google Scholar] [CrossRef]
- Ye, T.; Cheng, F.; Zhang, J.; Liu, Y.Z.; Wang, Q.; Deng, W.P. Highly Regioselective C–H Carbonylation of Alkenes with Phenyl Formate via Aryl to Vinyl 1,4-Palladium Migration. Org. Chem. Front. 2023, 10, 1537–1543. [Google Scholar] [CrossRef]
- Cacchi, S.; Fabrizi, G.; Goggiamani, A. Palladium-Catalyzed Hydroxycarbonylation of Aryl and Vinyl Halides or Triflates by Acetic Anhydride and Formate Anions. Org. Lett. 2003, 5, 4269–4272. [Google Scholar] [CrossRef]
- Korsager, S.; Taaning, R.H.; Skrydstrup, T. Effective Palladium-Catalyzed Hydroxycarbonylation of Aryl Halides with Substoichiometric Carbon Monoxide. J. Am. Chem. Soc. 2013, 135, 2891–2894. [Google Scholar] [CrossRef]
- Qi, X.; Li, C.L.; Jiang, L.B.; Zhang, W.Q.; Wu, X.F. Palladium-Catalyzed Alkoxycarbonylation of Aryl Halides with Phenols Employing Formic Acid as the CO Source. Catal. Sci. Technol. 2016, 6, 3099–3107. [Google Scholar] [CrossRef]
- Wu, F.P.; Peng, J.B.; Meng, L.S.; Qi, X.; Wu, X.F. Palladium-Catalyzed Ligand-Controlled Selective Synthesis of Aldehydes and Acids from Aryl Halides and Formic Acid. ChemCatChem 2017, 9, 3121–3124. [Google Scholar] [CrossRef]
- Hussain, N.; Chhalodia, K.A.; Ahmed, A.; Mukherjee, D. Recent Advances in Metal-Catalyzed Carbonylation Reactions by Using Formic Acid as CO Surrogate. ChemistrySelect 2020, 5, 11272–11290. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Ma, M.; Zhao, B. Palladium-Catalyzed Synthesis of Esters from Arenes through C–H Thianthrenation. Org. Lett. 2022, 24, 6031–6036. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Krasnova, L.; Finn, M.G.; Sharpless, K.B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed. 2014, 53, 9430–9448. [Google Scholar] [CrossRef] [PubMed]
- Revathi, L.; Ravindar, L.; Leng, J.; Rakesh, K.P.; Qin, H.L. Synthesis and Chemical Transformations of Fluorosulfates. Asian J. Org. Chem. 2018, 7, 662–682. [Google Scholar] [CrossRef]
- Veryser, C.; Demaerel, J.; Bieliunas, V.; Gilles, P.; De Borggraeve, W.M. Ex Situ Generation of Sulfuryl Fluoride for the Synthesis of Aryl Fluorosulfates. Org. Lett. 2017, 19, 5244–5247. [Google Scholar] [CrossRef]
- Guo, T.; Meng, G.; Zhan, X.; Yang, Q.; Ma, T.; Xu, L.; Sharpless, K.B.; Dong, J. A New Portal to SuFEx Click Chemistry: A Stable Fluorosulfuryl Imidazolium Salt Emerging as an “F-SO2+” Donor of Unprecedented Reactivity, Selectivity, and Scope. Angew. Chem. Int. Ed. 2018, 57, 2605–2610. [Google Scholar] [CrossRef]
- Zhou, H.; Mukherjee, P.; Liu, R.; Evrard, E.; Wang, D.; Humphrey, J.M.; Butler, T.W.; Hoth, L.R.; Sperry, J.B.; Sakata, S.K.; et al. Introduction of a Crystalline, Shelf-Stable Reagent for the Synthesis of Sulfur(VI) Fluorides. Org. Lett. 2018, 20, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Huang, L.; Zhou, P.; Xi, H.; Hu, J.; Zuo, Z.; Feng, H. Selectivity Controlled Hydroamination of Alkynes to Sulfonyl Fluoride Hubs: Development and Application. J. Org. Chem. 2022, 87, 4998–5004. [Google Scholar] [CrossRef]
- Gao, B.; Zhang, L.; Zheng, Q.; Zhou, F.; Klivansky, L.M.; Lu, J.; Liu, Y.; Dong, J.; Wu, P.; Sharpless, K.B. Bifluoride-Catalysed Sulfur(VI) Fluoride Exchange Reaction for the Synthesis of Polysulfates and Polysulfonates. Nat. Chem. 2017, 9, 1083–1088. [Google Scholar] [CrossRef]
- Dong, J.; Sharpless, K.B.; Kwisnek, L.; Oakdale, J.S.; Fokin, V.V. SuFEx-Based Synthesis of Polysulfates. Angew. Chem. Int. Ed. 2014, 53, 9466–9470. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.; Li, S.; Li, G.; Sharpless, K.B.; Wu, P. SuFEx Click Chemistry Enabled Late-Stage Drug Functionalization. J. Am. Chem. Soc. 2018, 140, 2919–2925. [Google Scholar] [CrossRef]
- Schimler, S.D.; Cismesia, M.A.; Hanley, P.S.; Froese, R.D.J.; Jansma, M.J.; Bland, D.C.; Sanford, M.S. Nucleophilic Deoxyfluorination of Phenols via Aryl Fluorosulfonate Intermediates. J. Am. Chem. Soc. 2017, 139, 1452–1455. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.; Tan, T.; Gu, Y.; Zhang, S.; Li, J.; Wang, Y.; Hou, W.; Yang, G.; Ma, P.; et al. Palladium-Catalyzed One-Pot Phosphorylation of Phenols Mediated by Sulfuryl Fluoride. Chem. Commun. 2021, 57, 4588–4591. [Google Scholar] [CrossRef] [PubMed]
- Hanley, P.S.; Clark, T.P.; Krasovskiy, A.L.; Ober, M.S.; O’Brien, J.P.; Staton, T.S. Palladium- and Nickel-Catalyzed Amination of Aryl Fluorosulfonates. ACS Catal. 2016, 6, 3515–3519. [Google Scholar] [CrossRef]
- Zhao, C.; Fang, W.Y.; Rakesh, K.P.; Qin, H.L. Pd-Catalyzed One-Pot Dehydroxylative Coupling of Phenols with K4[Fe(CN)6] Mediated by SO2F2: A Practical Method for the Direct Conversion of Phenols to Aryl Nitriles. Org. Chem. Front. 2018, 5, 1835–1839. [Google Scholar] [CrossRef]
- Fang, W.Y.; Leng, J.; Qin, H.L. SO2F2-Mediated One-Pot Synthesis of Aryl Carboxylic Acids and Esters from Phenols through a Pd-Catalyzed Insertion of Carbon Monoxide. Chem. Asian J. 2017, 12, 2323–2331. [Google Scholar] [CrossRef]
- Roth, G.P.; Thomas, J.A. Alkoxycarbonylation Reactions Using Aryl Fluorosulfonates. Tetrahedron Lett. 1992, 33, 1959–1962. [Google Scholar] [CrossRef]
- McGuire, M.A.; Sorenson, E.; Owings, F.W.; Resnick, T.M.; Fox, M.; Baine, N.H. A Novel, Practical Synthesis of Estra-1,3,5(10)-triene-3,17β-dicarboxylic Acid 17-tert-Butylamide (SK&F 105656) from Estrone, via a Palladium-Catalyzed Methoxycarbonylation of a 3-Fluorosulfonate. J. Org. Chem. 1994, 59, 6683–6686. [Google Scholar]
- Domino, K.; Veryser, C.; Wahlqvist, B.A.; Gaardbo, C.; Neumann, K.T.; Daasbjerg, K.; De Borggraeve, W.M.; Skrydstrup, T. Direct Access to Aryl Bis(trifluoromethyl)carbinols from Aryl Bromides or Fluorosulfates: Palladium-Catalyzed Carbonylation. Angew. Chem. Int. Ed. 2018, 57, 6858–6862. [Google Scholar] [CrossRef]
- Kockinger, M.; Hanselmann, P.; Hu, G.; Hone, C.A.; Kappe, C.O. Continuous Flow Synthesis of Aryl Aldehydes by Pd-Catalyzed Formylation of Phenol-Derived Aryl Fluorosulfonates Using Syngas. RSC Adv. 2020, 10, 22449–22453. [Google Scholar] [CrossRef]
- Ma, C.; Zhao, C.Q.; Xu, X.T.; Li, Z.M.; Wang, X.Y.; Zhang, K.; Mei, T.S. Nickel-Catalyzed Carboxylation of Aryl and Heteroaryl Fluorosulfates Using Carbon Dioxide. Org. Lett. 2019, 21, 2464–2467. [Google Scholar] [CrossRef] [PubMed]
- Song, X.D.; Guo, M.M.; Xu, S.; Shen, C.; Zhou, X.; Chu, X.Q.; Ma, M.; Shen, Z.L. Nickel-Catalyzed Diastereoselective Reductive Cross-Coupling of Disubstituted Cycloalkyl Iodides with Aryl Iodides. Org. Lett. 2021, 23, 5118–5122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ma, N.N.; Yu, Z.L.; Shen, C.; Zhou, X.; Chu, X.Q.; Rao, W.; Shen, Z.L. Palladium-Catalyzed Direct Reductive Cross-Coupling of Aryltrimethylammonium Salts with Aryl Bromides. Org. Chem. Front. 2021, 8, 4865–4870. [Google Scholar] [CrossRef]
- Cui, Y.Y.; Li, W.X.; Ma, N.N.; Shen, C.; Zhou, X.; Chu, X.Q.; Rao, W.; Shen, Z.L. Nickel-Catalyzed Direct Cross-Coupling of Heterocyclic Phosphonium Salts with Aryl Bromides. Org. Chem. Front. 2021, 8, 6931–6936. [Google Scholar] [CrossRef]
- Ma, N.N.; Ren, J.A.; Liu, X.; Chu, X.Q.; Rao, W.; Shen, Z.L. Nickel-Catalyzed Direct Cross-Coupling of Aryl Sulfonium Salt with Aryl Bromide. Org. Lett. 2022, 24, 1953–1957. [Google Scholar] [CrossRef]
- Li, W.X.; Yang, B.W.; Ying, X.; Zhang, Z.W.; Chu, X.Q.; Zhou, X.; Ma, M.; Shen, Z.L. Nickel-Catalyzed Direct Cross-Coupling of Diaryl Sulfoxide with Aryl Bromide. J. Org. Chem. 2022, 87, 11899–11908. [Google Scholar] [CrossRef]
- Ma, N.N.; Hu, X.B.; Wu, Y.S.; Zheng, Y.W.; Ma, M.; Chu, X.Q.; Xu, H.; Luo, H.; Shen, Z.L. Nickel-Catalyzed Direct Cross-Coupling of Aryl Thioether with Aryl Bromide. Org. Lett. 2023, 25, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Na, J.H.; Liu, X.; Jing, J.W.; Wang, J.; Chu, X.Q.; Ma, M.; Xu, H.; Zhou, X.; Shen, Z.L. Nickel-Catalyzed Direct Cross-Coupling of Aryl Fluorosulfates with Aryl Bromides. Org. Lett. 2023, 25, 2318–2322. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.A.; Na, J.H.; Gui, C.; Miao, C.; Chu, X.Q.; Ma, M.; Xu, H.; Zhou, X.; Shen, Z.L. Nickel-Catalyzed Direct Cross-Coupling of Unactivated Aryl Fluorides with Aryl Bromides. Org. Lett. 2023, 25, 5525–5529. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.A.; Chen, X.; Gui, C.; Miao, C.; Chu, X.Q.; Xu, H.; Zhou, X.; Ma, M.; Shen, Z.L. Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Phosphates with Aryl Bromides. Adv. Synth. Catal. 2023, 365, 2511–2515. [Google Scholar] [CrossRef]
- Liu, X.; He, C.Y.; Yin, H.N.; Miao, C.; Chu, X.Q.; Rao, W.; Xu, H.; Zhou, X.; Shen, Z.L. Nickel-Catalyzed Cross-Electrophile Coupling of Triazine Esters with Aryl Bromides. Chin. J. Chem. 2023, 41, 3539–3546. [Google Scholar] [CrossRef]
- Xu, H.; He, C.Y.; Huo, B.J.; Jing, J.W.; Miao, C.; Rao, W.; Chu, X.Q.; Zhou, X.; Shen, Z.L. Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Thiols with Aryl Bromides via C–S Bond Activation. Org. Chem. Front. 2023, 10, 5171–5179. [Google Scholar] [CrossRef]
- Wang, Q.D.; Liu, X.; Zheng, Y.W.; Wu, Y.S.; Zhou, X.; Yang, J.M.; Shen, Z.L. Iron-Mediated Reductive Amidation of Triazine Esters with Nitroarenes. Org. Lett. 2024, 26, 416–420. [Google Scholar] [CrossRef]
- Li, W.X.; Huo, B.J.; Huang, J.Y.; Rao, W.; Xu, H.; Zhou, X.; Shen, Z.L. Nickel-Catalyzed Cross-Electrophile Coupling of Unactivated (Hetero)aryl Sulfone with Aryl Bromide. J. Catal. 2024, 430, 115359. [Google Scholar] [CrossRef]
- Liu, C.; Yang, C.; Hwang, S.; Ferraro, S.M.; Flynn, J.P.; Niu, J. A General Approach to O-Sulfation by a Sulfur(VI) Fluoride Exchange Reaction. Angew. Chem. Int. Ed. 2020, 59, 18435–18441. [Google Scholar] [CrossRef]
- Liu, F.; Sohail, A.; Ablajan, K. Metal-Free Oxidative Formation of Aryl Esters by Catalytic Coupling of Acyl and Sulfonyl Chlorides with Arylboronic Acids. J. Org. Chem. 2024, 89, 27–33. [Google Scholar] [CrossRef]
- Iizumi, K.; Kurosawa, M.B.; Isshiki, R.; Muto, K.; Yamaguchi, J. Decarbonylative Synthesis of Aryl Nitriles from Aromatic Esters and Organocyanides by a Nickel Catalyst. Synlett 2021, 32, 1555–1559. [Google Scholar]
- Tukacs, J.M.; Marton, B.; Albert, E.; Toth, I.; Mika, L.T. Palladium-Catalyzed Aryloxy- and Alkoxycarbonylation of Aromatic Iodides in γ-Valerolactone as Bio-Based Solvent. J. Organomet. Chem. 2020, 923, 121407–121415. [Google Scholar] [CrossRef]
- Li, L.; Song, F.; Zhong, X.; Wu, Y.D.; Zhang, X.; Chen, J.; Huang, Y. Ligand-Controlled C-O Bond Coupling of Carboxylic Acids and Aryl Iodides: Experimental and Computational Insights. Adv. Synth. Catal. 2020, 362, 126–132. [Google Scholar] [CrossRef]
- Chai, L.L.; Zhao, Y.H.; Young, D.J.; Lu, X.; Li, H.X. Ni(II)-Mediated Photochemical Oxidative Esterification of Aldehydes with Phenols. Org. Lett. 2022, 24, 6908–6913. [Google Scholar] [CrossRef]
- Jha, D.K.; Sakkani, N.; Zhao, J.C.G. Visible Light-Assisted Direct C-H Acyloxylation of Polycyclic Aromatic Hydrocarbons using Carboxylic Acids. Adv. Synth. Catal. 2023, 365, 1585–1590. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
| Entry | Catalyst | Yield (%) b |
| 1 | Fe(acac)3 | 0 |
| 2 | Co(acac)2 | 0 |
| 3 | Mn(acac)3 | 0 |
| 4 | Cr(acac)3 | 0 |
| 5 | Ni(acac)2 | 0 |
| 6 | Pd(acac)2 | 73 |
| 7 | PdCl2 | 39 |
| 8 | Pd(PPh3)2Cl2 | 28 |
| 9 | Pd(PPh3)4 | 67 |
| 10 | Pd2(dba)3 | 75 |
| 11 | Pd(OAc)2 | 75 |
| 12 | Pd(OAc)2 | 82 c (84) d |
| 13 | Pd(OAc)2 | 80 e |
| 14 | Pd(OAc)2 | 83 f |
| 15 | Pd(OAc)2 | 21 g |
| 16 | - | 0 c |
| 17 | Pd(OAc)2 | 0 c,h |
| 18 | Pd(OAc)2 | 0 i |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Liang, Y.; Wang, W.-W.; Miao, C.; Chu, X.-Q.; Rao, W.; Xu, H.; Zhou, X.; Shen, Z.-L. Palladium-Catalyzed Esterification of Aryl Fluorosulfates with Aryl Formates. Molecules 2024, 29, 1991. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091991
Chen X, Liang Y, Wang W-W, Miao C, Chu X-Q, Rao W, Xu H, Zhou X, Shen Z-L. Palladium-Catalyzed Esterification of Aryl Fluorosulfates with Aryl Formates. Molecules. 2024; 29(9):1991. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091991
Chicago/Turabian StyleChen, Xue, Yuan Liang, Wen-Wen Wang, Chengping Miao, Xue-Qiang Chu, Weidong Rao, Hao Xu, Xiaocong Zhou, and Zhi-Liang Shen. 2024. "Palladium-Catalyzed Esterification of Aryl Fluorosulfates with Aryl Formates" Molecules 29, no. 9: 1991. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091991