
Sc-Modified C3N4 Nanotubes for High-Capacity Hydrogen Storage: A Theoretical Prediction
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
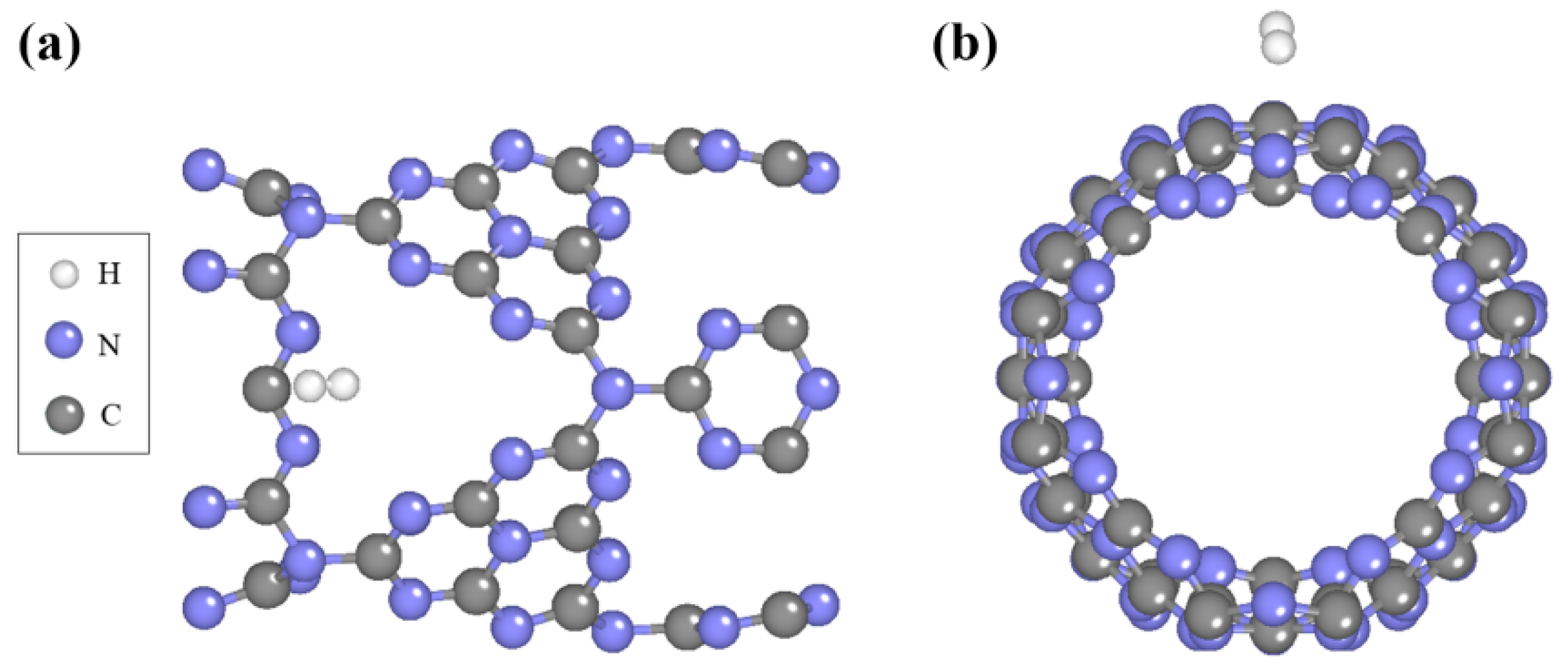
2.1. Geometric Structures of Pure C3N4 Nanotube and Single H2 Molecule Adsorption
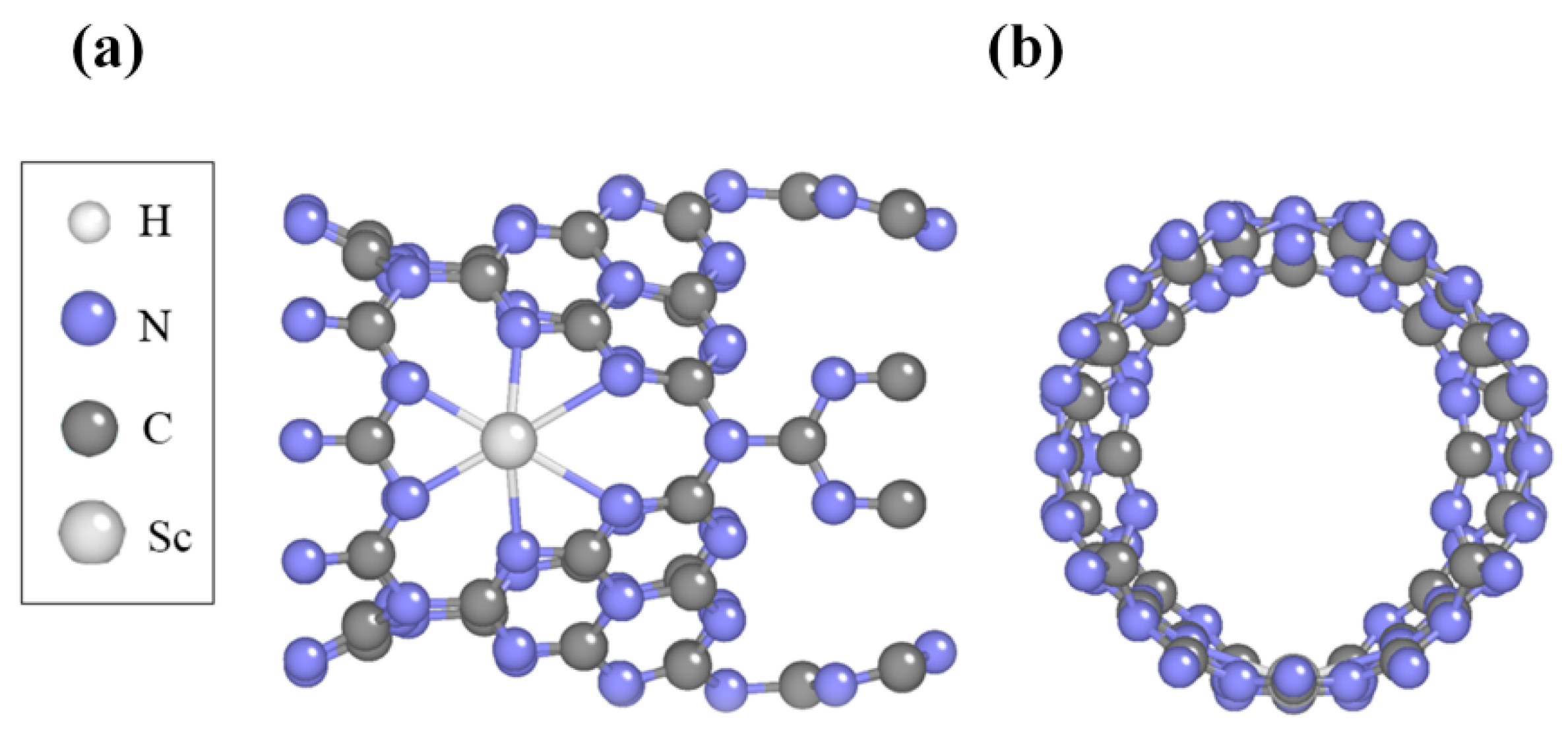
2.2. Structure and Stability of Sc-Modified C3N4 Nanotubes
2.3. H2 Molecules Adsorption on Sc-Modified C3N4 Nanotubes
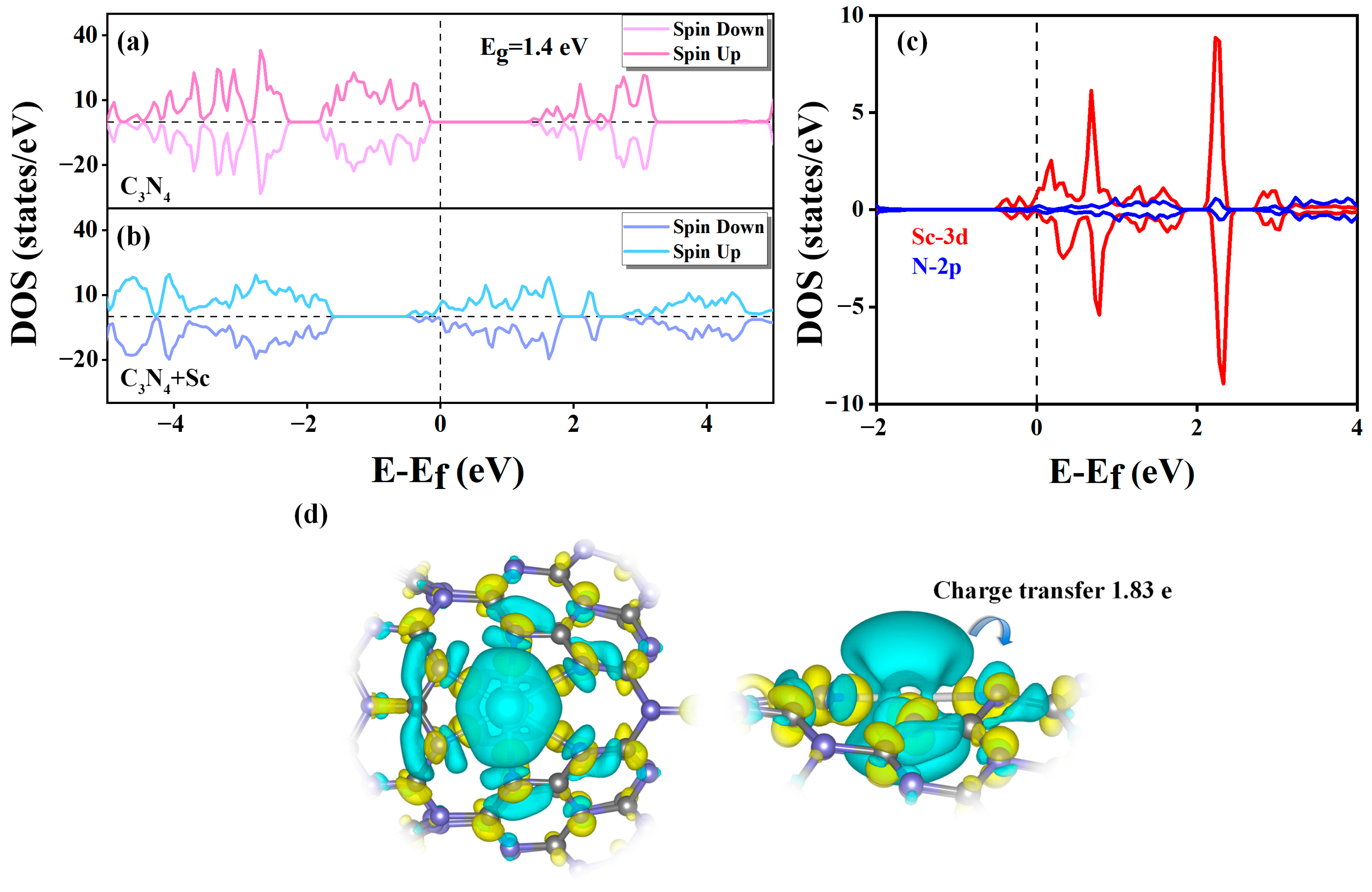
2.4. Interaction between H2 and Sc-Modified C3N4 Nanotube
2.5. Diffusion Energy Barrier for Hydrogen in a Tube
2.6. Molecule Dynamics for H2 Desorption
3. Computation Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, C.; Zhao, Q.; Zhang, G.; Xiong, B. Energy revolution: From a fossil energy era to a new energy era. Nat. Gas Ind. B 2016, 3, 1–11. [Google Scholar] [CrossRef]
- Ouyang, L.; Huang, J.; Fang, C.; Zhang, Q.; Sun, D.; Zhu, M. The controllable hydrolysis rate for LaMg12 hydride. Int. J. Hydrogen Energy 2012, 37, 12358–12364. [Google Scholar] [CrossRef]
- Dawood, F.; Anda, M.; Shafiullah, G.M. Hydrogen production for energy: An overview. Int. J. Hydrogen Energy 2020, 45, 3847–3869. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhu, X.; Gao, X.; An, C.; Wang, Z.; Zuo, C.; Dionysiou, D.D.; He, H.; Jiang, Z. Enhancing photocatalytic CO2 reduction with TiO2-based materials: Strategies, mechanisms, challenges, and perspectives. Environ. Sci. Ecotechnol. 2024, 20, 100368. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cao, L.; Chang, Y.; Yuan, Z.; Zhang, S.; Liu, S.; Zhang, M.; Fan, H.; Jiang, Z. Improving the CO2 Hydrogenation Activity of Photocatalysts via the Synergy between Surface Frustrated Lewis Pairs and the CuPt Alloy. ACS Sustain. Chem. Eng. 2023, 11, 5597–5607. [Google Scholar] [CrossRef]
- Zhu, X.; Zong, H.; Pérez, C.J.V.; Miao, H.; Sun, W.; Yuan, Z.; Wang, S.; Zeng, G.; Xu, H.; Jiang, Z. Supercharged CO2 photothermal catalytic methanation: High conversion, rate, and selectivity. Angew. Chem. 2023, 135, e202218694. [Google Scholar] [CrossRef]
- Kothari, R.; Buddhi, D.; Sawhney, R.L. Comparison of environmental and economic aspects of various hydrogen production methods. Renew. Sustain. Energy Rev. 2008, 12, 553–563. [Google Scholar] [CrossRef]
- Sinigaglia, T.; Lewiski, F.; Santos Martins, M.E.; Mairesse Siluk, J.C. Production, storage, fuel stations of hydrogen and its utilization in automotive applications-a review. Int. J. Hydrogen Energy 2017, 42, 24597–24611. [Google Scholar] [CrossRef]
- George, L.; Saxena, S.K. Structural stability of metal hydrides, alanates and borohydrides of alkali and alkali-earth elements: A review. Int. J. Hydrogen Energy 2010, 35, 5454–5470. [Google Scholar] [CrossRef]
- Jiang, Z.; Yuan, Z.; Duchesne, P.N.; Sun, W.; Lyu, X.; Miao, W.; Viasus Pérez, C.J.; Xu, Y.; Yang, D.; Huang, B.; et al. A living photocatalyst derived from CaCu3Ti4O12 for CO2 hydrogenation to methanol at atmospheric pressure. Chem. Catal. 2023, 3, 100507. [Google Scholar] [CrossRef]
- Tai, X.; Yan, X.; Wang, L. Synthesis, Structural Characterization, Hirschfeld Surface Analysis, Density Functional Theory, and Photocatalytic CO2 Reduction Activity of a New Ca(II) Complex with a Bis-Schiff Base Ligand. Molecules 2024, 29, 1047. [Google Scholar] [CrossRef]
- Wang, L.H.; Tai, X.S. Synthesis, Structural Characterization, Hirschfeld Surface Analysis and Photocatalytic CO2 Reduction Activity of a New Dinuclear Gd(III) Complex with 6-Phenylpyridine-2-Carboxylic Acid and 1,10-Phenanthroline Ligands. Molecules 2023, 28, 7595. [Google Scholar] [CrossRef]
- Yu, X.; Wu, Z.; Xia, B.; Xu, N. Enhancement of hydrogen storage capacity of Ti–V–Cr–Mn BCC phase alloys. J. Alloys Compd. 2004, 372, 272–277. [Google Scholar] [CrossRef]
- Wong-Foy, A.G.; Matzger, A.J.; Yaghi, O.M. Exceptional H2 Saturation Uptake in Microporous Metal−Organic Frameworks. J. Am. Chem. Soc. 2006, 128, 3494–3495. [Google Scholar] [CrossRef]
- Kudlek, E.; Dudziak, M.; Bohdziewicz, J. Influence of inorganic ions and organic substances on the degradation of pharmaceutical compound in water matrix. Water 2016, 8, 532. [Google Scholar] [CrossRef]
- Züttel, A.; Remhof, A.; Borgschulte, A.; Friedrichs, O. Hydrogen: The future energy carrier. Philos. Trans. R. Soc. A 2010, 368, 3329–3342. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Myers, A.L. Optimum conditions for adsorptive storage. Langmuir 2006, 22, 1688–1700. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Q.; Sun, Q.; Jena, P.; Chen, X. Electric field enhanced hydrogen storage on polarizable materials substrates. Proc. Natl. Acad. Sci. USA 2010, 107, 2801–2806. [Google Scholar] [CrossRef]
- Ju, L.; Liu, J.; Wang, M.; Yang, S.; Liu, S. Modulation of charge in C9N4 monolayer for a high-capacity hydrogen storage as a switchable strategy. Front. Phys. 2024, 19, 43208. [Google Scholar] [CrossRef]
- Isidro-Ortega, F.J.; Pacheco-Sánchez, J.H.; Desales-Guzmán, L.A. Hydrogen storage on lithium decorated zeolite templated carbon, DFT study. Int. J. Hydrogen Energy 2017, 42, 30704–30717. [Google Scholar] [CrossRef]
- Energy (US-DOE) Standard. Available online: https://www.standards.doe.gov/ (accessed on 2 June 2023).
- Gao, F.; Sun, J.T.; Meng, S. “H2 sponge”: Pressure as a means for reversible high-capacity hydrogen storage in nanoporous Ca-intercalated covalent organic frameworks. Nanoscale 2015, 7, 6319–6324. [Google Scholar] [CrossRef]
- Bellosta von Colbe, J.; Ares, J.-R.; Barale, J.; Baricco, M.; Buckley, C.; Capurso, G.; Gallandat, N.; Grant, D.M.; Guzik, M.N.; Jacob, I.; et al. Application of hydrides in hydrogen storage and compression: Achievements, outlook and perspectives. Int. J. Hydrogen Energy 2019, 44, 7780–7808. [Google Scholar] [CrossRef]
- Heung, L.K. Using Metal Hydride to Store Hydrogen; Department of Energy: Washington, DC, USA, 2003.
- Sakintuna, B.; Lamari-Darkrim, F.; Hirscher, M. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen Energy 2007, 32, 1121–1140. [Google Scholar] [CrossRef]
- Zaluski, L.; Zaluska, A.; Tessier, P.; Ström-Olsen, J.; Schulz, R. Effects of relaxation on hydrogen absorption in Fe-Ti produced by ball-milling. J. Alloys Compd. 1995, 227, 53–57. [Google Scholar] [CrossRef]
- Floriano, R.; Leiva, D.R.; Dessi, J.G.; Asselli, A.A.C.; Jorge Junior, A.M.; Botta, W.J. Mg-based nanocomposites for hydrogen storage containing Ti-Cr-V alloys as additives. Mater. Res. 2016, 19, 80–85. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.K.; Srivastava, O. On the synthesis of the Mg2Ni alloy by mechanical alloying. J. Alloys Compd. 1995, 227, 63–68. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef]
- Kleperis, J.; Lesnicenoks, P.; Grinberga, L.; Chikvaidze, G.; Klavins, J. Zeolite as material for hydrogen storage in transport applications/CEOLĪTA KĀ ŪDEŅRAŽA UZGLABĀŠANAS VIDES IZPĒTE. Latv. J. Phys. Tech. Sci. 2013, 50, 59–64. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Z.; Zhu, Y. Porous carbon-based materials for hydrogen storage: Advancement and challenges. J. Mater. Chem. A 2013, 1, 9365–9381. [Google Scholar] [CrossRef]
- Sakintuna, B.; Yürüm, Y. Templated porous carbons: A review article. Ind. Eng. Chem. Res. 2005, 44, 2893–2902. [Google Scholar] [CrossRef]
- Chen, Z.; Kirlikovali, K.O.; Idrees, K.B.; Wasson, M.C.; Farha, O.K. Porous materials for hydrogen storage. Chem 2022, 8, 693–716. [Google Scholar] [CrossRef]
- Roongcharoen, T.; Impeng, S.; Chitpakdee, C.; Rungrotmongkol, T.; Jitwatanasirikul, T.; Jungsuttiwong, S.; Namuangruk, S. Intrinsic property and catalytic performance of single and double metal atoms incorporated g-C3N4 for O2 activation: A DFT insight. Appl. Surf. Sci. 2021, 541, 148671. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Wagh, V.; Rout, C.S.; Chakraborty, B. High capacity reversible hydrogen storage in zirconium doped 2D-covalent triazine frameworks: Density Functional Theory investigations. Int. J. Hydrogen Energy 2021, 46, 14520–14531. [Google Scholar] [CrossRef]
- Mahamiya, V.; Shukla, A.; Chakraborty, B. Scandium decorated C24 fullerene as high capacity reversible hydrogen storage material: Insights from density functional theory simulations. Appl. Surf. Sci. 2022, 573, 151389. [Google Scholar] [CrossRef]
- Chen, P.; Wu, X.; Lin, J.; Tan, K.L. High H2 Uptake by Alkali-Doped Carbon Nanotubes Under Ambient Pressure and Moderate Temperatures. Science 1999, 285, 91–93. [Google Scholar] [CrossRef]
- Wu, M.; Wang, Q.; Sun, Q.; Jena, P. Functionalized graphitic carbon nitride for efficient energy storage. J. Phys. Chem. C 2013, 117, 6055–6059. [Google Scholar] [CrossRef]
- Sun, Z.; Lu, X.; Nyahuma, F.M.; Yan, N.; Xiao, J.; Su, S.; Zhang, L. Enhancing hydrogen storage properties of MgH2 by transition metals and carbon materials: A brief review. Front. Chem. 2020, 8, 552. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, Y.; Wu, W.; Jiang, L.; Dong, S. Hydrogen storage of capped single-walled carbon nanotube via transition-metal doping. Europhys. Lett. 2013, 104, 36002. [Google Scholar] [CrossRef]
- Alex, K.V.; Prabhakaran, A.; Jayakrishnan, A.; Kamakshi, K.; Silva, J.P.B.; Sekhar, K. Charge coupling enhanced photocatalytic activity of BaTiO3/MoO3 heterostructures. ACS Appl. Mater. Interfaces 2019, 11, 40114–40124. [Google Scholar] [CrossRef]
- Panigrahi, P.; Kumar, A.; Karton, A.; Ahuja, R.; Hussain, T. Remarkable improvement in hydrogen storage capacities of two-dimensional carbon nitride (g-C3N4) nanosheets under selected transition metal doping. Int. J. Hydrogen Energy 2020, 45, 3035–3045. [Google Scholar] [CrossRef]
- Stoyanov, S.R.; Titov, A.V.; Král, P. Transition metal and nitrogen doped carbon nanostructures. Coord. Chem. Rev. 2009, 253, 2852–2871. [Google Scholar] [CrossRef]
- Durgun, E.; Ciraci, S.; Yildirim, T. Functionalization of carbon-based nanostructures with light transition-metal atoms for hydrogen storage. Phys. Rev. B 2008, 77, 085405. [Google Scholar] [CrossRef]
- Nachimuthu, S.; Lai, P.-J.; Jiang, J.-C. Efficient hydrogen storage in boron doped graphene decorated by transition metals—A first-principles study. Carbon 2014, 73, 132–140. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Q.; Jena, P.; Kawazoe, Y. Clustering of Ti on a C60 surface and its effect on hydrogen storage. J. Am. Chem. Soc. 2005, 127, 14582–14583. [Google Scholar] [CrossRef]
- Nisha, V.; Moolayadukkam, S.; Paravannoor, A.; Panoth, D.; Chang, Y.-H.; Palantavida, S.; Hinder, S.J.; Pillai, S.C.; Vijayan, B.K. Cu doped graphitic C3N4 for p-nitrophenol reduction and sensing applications. Inorg. Chem. Commun. 2022, 142, 109598. [Google Scholar] [CrossRef]
- Das, T.K.; Banerjee, S.; Vishwanadh, B.; Joshi, R.; Sudarsan, V. On the nature of interaction between Pd nanoparticles and C3N4 support. Solid State Sci. 2018, 83, 70–75. [Google Scholar] [CrossRef]
- Guan, P.; Yang, B.; Liu, J.; Yin, H.; Jiang, J.; Sui, L.; Yang, S. Synthesis of novel rare-earth cerium doped C3N4 nanocomposites for boosting photocatalytic H2 evolution. Chem. Phys. Lett. 2023, 811, 140222. [Google Scholar] [CrossRef]
- Dong, S.; Lv, E.; Wang, J.; Li, C.; Ma, K.; Gao, Z.; Yang, W.; Ding, Z.; Wu, C.; Gates, I.D. Construction of transition metal-decorated boron doped twin-graphene for hydrogen storage: A theoretical prediction. Fuel 2021, 304, 121351. [Google Scholar] [CrossRef]
- Luo, Z.; Fan, X.; Pan, R.; An, Y. A first-principles study of Sc-decorated graphene with pyridinic-N defects for hydrogen storage. Int. J. Hydrogen Energy 2017, 42, 3106–3113. [Google Scholar] [CrossRef]
- Hongzhiwei Technology, D.S., Version V2023A, China. 2021. Available online: https://iresearch.net.cn/cloudSoftware (accessed on 2 June 2023).
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Chaudhuri, S.; Graetz, J.; Ignatov, A.; Reilly, J.J.; Muckerman, J.T. Understanding the role of Ti in reversible hydrogen storage as sodium alanate: A combined experimental and density functional theoretical approach. J. Am. Chem. Soc. 2006, 128, 11404–11415. [Google Scholar] [CrossRef]
- Xiong, R.; Sang, G.; Zhang, G.; Yan, X.; Li, P.; Yao, Y.; Luo, D.; Chen, C.A.; Tang, T. Evolution of the active species and catalytic mechanism of Ti doped NaAlH4 for hydrogen storage. Int. J. Hydrogen Energy 2017, 42, 6088–6095. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, B.; Wu, Y.; Lv, W.; Zhou, S. Theoretical prediction and experimental study on catalytic mechanism of incorporated Ni for hydrogen absorption of Mg. Int. J. Hydrogen Energy 2019, 44, 27885–27895. [Google Scholar] [CrossRef]
- Han, Z.; Yeboah, M.L.; Jiang, R.; Li, X.; Zhou, S. Hybrid activation mechanism of thermal annealing for hydrogen storage of magnesium based on experimental evidence and theoretical validation. Appl. Surf. Sci. 2020, 504, 144491. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Li, D.-H.; Li, Q.-M.; Qi, S.-L.; Qin, H.-C.; Liang, X.-Q.; Li, L. Theoretical Study of Hydrogen Production from Ammonia Borane Catalyzed by Metal and Non-Metal Diatom-Doped Cobalt Phosphide. Molecules 2022, 27, 8206. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Sheng, L. Al-Decorated C2N Monolayer as a Potential Catalyst for NO Reduction with CO Molecules: A DFT Investigation. Molecules 2022, 27, 5790. [Google Scholar] [CrossRef]
- Li, X.; Dai, Y.; Ma, Y.; Li, M.; Yu, L.; Huang, B. Landscape of DNA-like inorganic metal free double helical semiconductors and potential applications in photocatalytic water splitting. J. Mater. Chem. A 2017, 5, 8484–8492. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Zhang, X.; Yang, Y.; Gao, M.; Pan, H. Superior catalytic activity derived from a two-dimensional Ti3C2 precursor towards the hydrogen storage reaction of magnesium hydride. Chem. Commun. 2016, 52, 705. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, Z.; Jian, N.; Hu, J.; Du, F.; Yao, J.; Gao, M.; Liu, Y.; Pan, H. A novel complex oxide TiVO3.5 as a highly active catalytic precursor for improving the hydrogen storage properties of MgH2. Int. J. Hydrogen Energ. 2018, 43, 23327–23335. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, Z.; Zhang, M.; Gao, M.; Hu, J.; Du, F.; Liu, Y.; Pan, H. A novel solid-solution MXene (Ti0.5V0.5)3C2 with high catalytic activity for hydrogen storage in MgH2. Materialia 2018, 1, 114–120. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, H.; Zhu, Y.; Li, S.; Zhang, J.; Li, L. Excellent catalytic activity of a two-dimensional Nb4C3Tx (MXene) on hydrogen storage of MgH2. Appl. Surf. Sci. 2019, 493, 431–440. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Tang, X.; He, C.; Wang, T.; Shang, L.; Wang, M.; Yang, S.; Tang, Z.; Ju, L. Sc-Modified C3N4 Nanotubes for High-Capacity Hydrogen Storage: A Theoretical Prediction. Molecules 2024, 29, 1966. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091966
Liu S, Tang X, He C, Wang T, Shang L, Wang M, Yang S, Tang Z, Ju L. Sc-Modified C3N4 Nanotubes for High-Capacity Hydrogen Storage: A Theoretical Prediction. Molecules. 2024; 29(9):1966. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091966
Chicago/Turabian StyleLiu, Shuli, Xiao Tang, Chang He, Tingting Wang, Liying Shang, Mengyuan Wang, Shenbo Yang, Zhenjie Tang, and Lin Ju. 2024. "Sc-Modified C3N4 Nanotubes for High-Capacity Hydrogen Storage: A Theoretical Prediction" Molecules 29, no. 9: 1966. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091966