Y(OTf)3-Salazin-Catalyzed Asymmetric Aldol Condensation


State Key Laboratory of Chemical Resource Engineering, Department of Organic Chemistry, College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, China
*
Author to whom correspondence should be addressed.
Molecules 2024, 29(9), 1963; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091963
Submission received: 2 April 2024
/
Revised: 21 April 2024
/
Accepted: 22 April 2024
/
Published: 25 April 2024
(This article belongs to the Section Organic Chemistry)
Abstract
:The chiral aziridine-containing vicinal iminophenol tridentate ligands (named salazins) are a class of readily prepared chiral ligands from enantiopure aziridines and salicylaldehydes. Their scandium and yttrium triflate complexes show excellent reactivity and enantioselectivities in the catalytic asymmetric aldol condensation of electron-deficient aromatic aldehydes and ketones, including acetone and cycloalkanones. The stereoselectivity is rationalized to the strong π–stacking interaction between aromatic aldehydes and the vicinal iminophenol group in the chiral ligands.
1. Introduction
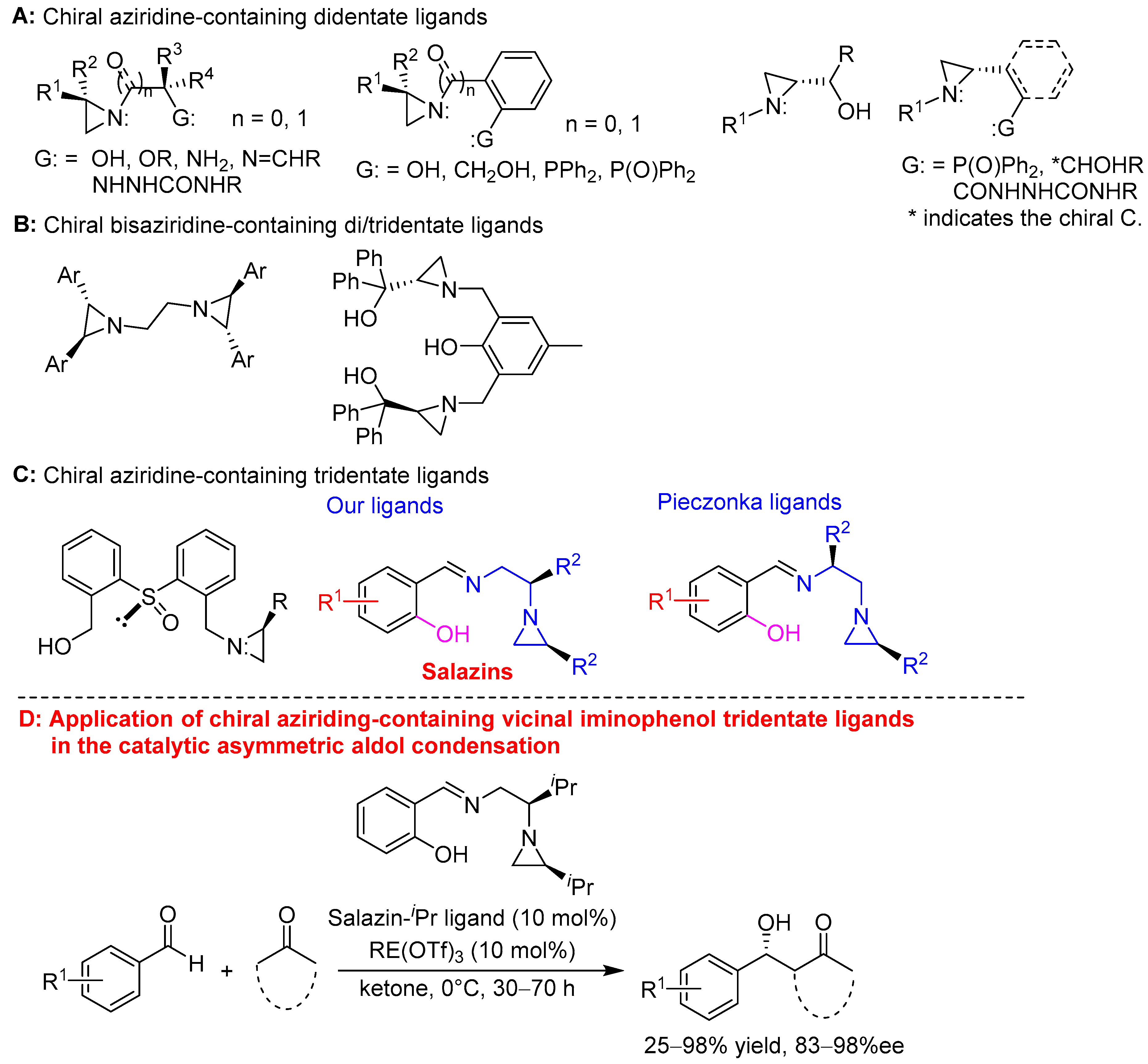
Aziridine-containing chiral ligands are an important class of chiral ligands in asymmetric catalysis and have been applied in various asymmetric transformations [1,2] because enantiopure aziridines are easily prepared chiral resources from commercially available vicinal amino alcohols [3,4]. Most developed aziridine-containing chiral ligands are chiral bidentate ligands, including chiral aziridine-containing alcohols [5,6,7], ethers [8,9,10], phenols [11], amines [12], imines [13], semicarbazides [14,15], phosphines [16,17], phosphine oxides [18] (Scheme 1A), and aziridines, which are actually bis-aziridine ligands [19,20,21,22] (Scheme 1B. alternatively, multidentate phenol-derived bis-aziridine-2-methanol chiral ligand has also developed [5] (Scheme 1B)). However, only limited aziridine-containing chiral tridentate ligands have been designed and synthesized, for instance, 2-[2-(aziridine-1-methyl)benzenesulfinyl]benzyl alcohols [23,24,25] and two classes of aziridine-containing vicinal iminophenol tridentate ligands [13,26] (Scheme 1C). Although various chiral aziridine-containing ligands have been prepared and applied in several asymmetric catalysis, their applications are still limited to date due to their complicated and multiple synthetic steps. To enrich the aziridine-containing chiral ligands, recently, our research group synthesized a series of chiral aziridine-containing vicinal iminophenol tridentate ligands in one step from readily prepared enantiopure aziridines and salicylaldehydes. The regiospecific cleavage of the more substituted C–N bond of aziridines was enabled by an iminium-mediated self-ring opening reaction with the complete inversion of configuration, because nucleophilic ring-opening is an electronic effect-controlled process (Scheme 1C, middle ones) [26,27]. In short, our tridentate ligands were named salazins because they were prepared directly from salicylaldehydes and aziridines. They exhibited good to excellent enantioselectivity in the Zn(OTf)2-catalyzed asymmetric aldol condensation [26].
Lewis acids generally play an important role in their unique catalytic reactivity and selectivity in catalytic organic reactions. In comparison with traditional Lewis acids of transition metal ions, rare-earth metal ions generally show strong Lewis acidity and oxophilicity. The rare-earth metal triflates are more appealing from a synthetic point of view in acid-/Lewis acid-catalyzed organic reactions. In the past three decades, rare-earth metal triflates, including scandium triflate, yttrium triflate, and lanthanide (La–Lu) triflates, etc., have been widely utilized in the organic chemistry [28,29]. China has abundant rare-earth metal resources. It is in demand to develop their application in catalytic organic reactions, especially catalytic asymmetric organic reactions.
2. Results and Discussion
When we first prepared our chiral aziridine-containing vicinal iminophenol tridentate ligands (salazins), they were evaluated with a catalytic asymmetric aldol condensation of aromatic aldehydes and acetone in the presence of Zn(OTf)2, affording β-hydroxyketones in good to excellent yields and enantioselectivities [26]. Because the rare-earth metal ions generally show stronger Lewis acidity and oxophilicity than transition metal ions [28], we rationalized that rare-earth metal triflates would take advantage in the Lewis acid-catalyzed aldol condensation. To develop the further application of our chiral tridentate ligands and rare-earth metals, we conducted a rare-earth metal triflate chiral tridentate ligand complex-catalyzed asymmetric aldol condensation. The reaction of 4-nitrobenzaldehyde (1a) and acetone was selected as a model reaction to optimize reaction conditions. The solvent screening was first carried out with the Sc(OTf)3-salazin-Bn complex as the catalyst (Table 1). The ligand salazin-Bn was prepared from salicylaldehyde and (S)-benzylaziridine. When toluene was used as solvent, the aldol adduct 3a was obtained in only a 3% yield with 38% ee (Table 1, entry 1). The adduct 3a was obtained in an excellent yield of 98%, but moderate 53% ee, in EtOH (Table 1, entry 2). When the solvent was changed to DCM and THF, the adduct was generated in 10% and 20% yields with 60% ee and 49% ee, respectively (Table 1, entries 3 and 4). Furthermore, an excellent yield of 96% and good enantioselectivity (87% ee) were obtained when the reaction was performed in acetone (Table 1, entry 5). The results indicated that acetone, in which 87% ee was obtained, was the best choice as the solvent for the asymmetric reaction.
For convenience, the chiral ligands and rare-earth metal triflates were further optimized in acetone with the model reaction at room temperature (15 °C) (Table 2, entries 1–14). The reaction of 4-nitrobenzaldehyde (1a) and acetone under the catalysis of Sc(OTf)3 and ligand salazin-Bn generated adduct 3a in a 96% yield with 81% ee (Table 2, entry 1). Under the catalysis of Sc(OTf)3 with ligands salazin-tBu-Bn and salazin-Br-Bn, the adduct 3a was obtained in a 96% yield with 70% ee and a 55% yield with 78% ee, respectively (Table 2, entries 2 and 3). The reaction gave adduct 3a in a 91% yield with 86% ee and an 89% yield with 87% ee, respectively, under the catalysis of Sc(OTf)3 with ligands salazin-NO2-iPr and salazin-Br-iPr (Table 2, entries 4 and 5). However, under the catalysis of Sc(OTf)3 with ligand salazin-I-iPr, both the yield (60%) and enantioselectivity (80% ee) of the adduct 3a decreased obviously (Table 2, entry 6). The reaction catalyzed by Sc(OTf)3 with unsubstituted ligand salazin-iPr produced adduct 3a in 98% with 88% ee, showing the best results (Table 2, entry 7). TLC monitoring the reaction mixture indicated that the aldehyde 1a disappeared completely for 26 h, revealing that the ligand salazin-iPr was more efficient. Thus, further screening of different rare-earth metal triflates was conducted with ligand salazin-iPr, and the reaction was stirred at 15 °C for 26 h. The Y(OTf)3-catalyzed reaction gave rise to the same results as the Sc(OTf)3-catalyzed one (Table 2, entry 8). Other rare-earth metal triflates, including La(OTf)3, Ce(OTf)3, Eu(OTf)3, Gd(OTf)3, and Lu(OTf)3, were also attempted. Although excellent yields (91–98%) and enantioselectivities (83–87% ee) were observed, all of them are lower than those in the Sc(OTf)3 and Y(OTf)3-catalyzed reactions (Table 2, entries 9–13). In comparison with the transition metal catalyst Zn(OTf)2, the model reaction was conducted under the catalysis of Zn(OTf)2 and salazin-iPr, giving adduct 3a in a 90% yield and 70% ee (Table 2, entry 14). Both the yield and enantioselectivity were lower than those in the tested rare-earth metal triflate-catalyzed reactions. Finally, the reaction temperature was optimized, as well (Table 2, entries 15–20). The reactions under the catalysis of Sc(OTf)3 and ligand salazin-iPr generated adduct 3a in the same excellent yield of 98%, but different good to excellent enantioselectivities, 85% ee at 40 °C, 88% ee at 20 °C, and 98% ee at 0 °C (Table 2, entries 15–17). Similarly, the reactions under the catalysis of Y(OTf)3 and ligand salazin-iPr gave adduct 3a in excellent yields of 96–98%, but different good to excellent enantioselectivities, 89% ee at 40 °C, 88% ee at 20 °C, and 98% ee at 0 °C (Table 2, entries 18–20). All the reaction times were determined by the results of TLC monitoring the reaction mixtures. The results reveal that the reaction rates increase along with the elevation of the reaction temperature. The results illustrate that both catalytic combinations of Sc(OTf)3 and Y(OTf)3 with ligand salazin-iPr at 0 °C are the best choice. However, considering that the metal scandium is more expensive than the metal yttrium, the catalytic system of Y(OTf)3 and ligand salazin-iPr was selected as the optimal combination in the asymmetric aldol condensation of aldehydes and ketones at 0 °C.
With the optimal conditions in hand, the scope of aldehydes 1 was examined (Scheme 2). Similar to 4-nitrobenzaldehyde (1a), strong electron-deficient 3-nitro and 2-nitrobenzaldehydes (1b and 1c) generated the corresponding adducts 3b and 3c in good yields of 72% and 84%, with excellent enantioselectivities of 90% ee and 93% ee. Both yields and enantioselectivities are lower than those of 4-nitrobenzaldehyde (1a). 4-Cyanobenzaldehydes (1d) led to the desired adduct 3d in an excellent yield (93%) and enantioselectivity (91% ee). Differently, weak electron-deficient 4-chloro- and 4-bromobenzaldehydes (1e and 1f) gave rise to the expected adducts 3e and 3f in low yields of 33% and 39%, but good to excellent enantioselectivities of 91% ee and 88% ee, respectively. However, weak electron-deficient 4-fluorobenzaldehyde (1g), benzaldehyde (1g), and electron-rich 4-methoxybenzaldehyde (1i) did not undergo the asymmetric reactions under the same reaction conditions, showing the electronic effect-dependent substrate selectivity possibly due to weak π-stacking interaction between these aromatic aldehydes and the benzene ring in the ligand backbone, similar to that in the catalytic asymmetric aziridination of chalcones [30]. Although the fluorine element has the strongest electronegativity, it presents weak electron-withdrawing ability in fluoroarenes, such as 4-fluorobenzaldehyde and 4-fluorobenzoic acid, because it exists in the same period with carbon in the periodic table. Its 2p orbital can overlap very well with the 2p orbital of the carbon atom in the arene ring, exhibiting strong electron-donating conjugative effect, which neutralizes its strong electron-withdrawing inductive effect. As a result, the fluoro group presents as a weak electron-withdrawing group. It can be seen from that the Hammett constant of F is smaller than those of Cl and Br, and the acidity of 4-fluorobenzoic acid is weaker than those of 4-chloro and 4-bromobenzoic acids. Furthermore, both bicyclic fused naphthalene-1-carbaldehyde (1j) and naphthalene-2-carbaldehyde (1k) proceeded well the reaction, affording the corresponding adducts 3j in a 35% yield with 96% ee and 3k in an 84% yield with 91% ee, respectively. In addition, bicyclic fused quinoline-4-carbaldehyde (1l) also worked well, giving the desired adduct 3l in an 89% yield with 89% ee. All bicyclic fused arene and heteroarenecarbaldehydes showed excellent reactivities and stereoselectivities, possibly due to the existence of strong π-stacking interaction between these (hetero)arenecarbaldehydes and the benzene ring in the ligand backbone. To further extend the scope of aldehydes, two functionalized benzaldehydes, 4-methanesulfonylbenzaldehyde (1m) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (1n), were examined, affording the expected adducts 3m in an 86% yield and 92% ee, with a dehydrated side product 4-(4-methanesulfonylphenyl)but-3-en-2-one (4m) 14% yield and 3n in a 70% yield and 87% ee, respectively. The aldehyde 1n required long reaction time of 70 h. The product 3n cannot be obtained under basic catalyzed asymmetric conditions due to the existence of base sensitive methanesulfonyl group. It is a useful synthetic building block in various organic transformations. To test the scope of aldehydes, aliphatic cyclohexanecarbaldehyde (1o) was attempted, but no reaction occurred due to the absence of the π-stacking interaction between the aldehyde and the benzene ring in the ligand backbone.
To investigate the scope of ketones, several cycloalkanones 2b–2d were evaluated in the reaction with 4-nitrobenzaldehyde (1a) under the catalysis of Y(OTf)3 and ligand salazin-iPr (Table 3). The results indicate that cyclobutanone (2b) exhibits poor reactivity (25% yield) and diastereoselectivity (anti:syn 53:47) but moderate to good enantioselectivities (83% ee and 69% ee) for the diastereomeric products anti-5a and syn-5a, respectively (Table 3, entry 1). However, the reactions of cyclopentanone (2c) and cyclohexanone (2d) show good to excellent diastereoselectivities (the ratio of anti:syn diastereomers 83:17 and 95:6, respectively) and enantioselectivities (96% ee and 91% ee) for the anti-diastereomeric products anti-5b and anti-5c, respectively (Table 3, entries 2 and 3). The stereostructures of anti- and syn-diastereomers were identified in comparison with the reported NMR data [31,32,33]. Linear pentan-3-one (2e) was also subjected to the catalytic asymmetric reaction with 4-nitrobenzaldehyde (1a) at 0 °C for 50 h, affording the desired product 5d in a low yield of 30% due to steric hindrance, with a diastereomeric ratio of anti:syn = 94:6 and enantioselectivities of 98% ee and 71% ee for anti-5d and syn-5d, respectively. The results indicate that anti-diastereomeric products generally show higher enantioselectivities than the corresponding syn-diastereomeric ones. Aromatic acetophenone (2f) was evaluated in the asymmetric catalysis, with aldehyde 1a, as well. The asymmetric reaction was conducted at room temperature (at 25 °C) for 30 h because the melting point of acetophenone is 20 °C. The expected product 5e was obtained in only a 15% yield with 44% ee. The asymmetric catalytic reaction of 1a and 2f was also performed in acetone as a solvent at 0 °C for 30 h. However, only aldol adduct 3a was obtained in a 98% yield and 98% ee. The results reveal that acetone is more active than acetophenone in the asymmetric catalytic aldol condensation with 4-nitrobenzaldehyde (1a) (Scheme 3).
On the basis of the reported coordination of Y(OTf)3 [29] and the absolute configuration of adducts 3, the plausible mechanism of the catalytic asymmetric reaction is proposed as follows. Tridentate ligand salazin-iPr first coordinates with Y(OTf)3 to form the complex A. Benzaldehyde (1a) coordinates with the central metal Y from the top side of the catalyst by exchange with triflate, generating a new complex B. The enolic form of acetone is generated under the acid catalysis and attacks the coordinated benzaldehyde in the complex B from the top side of benzaldehyde through a hydrogen bond-bound six-membered ring transition state TS. After the nucleophilic attack, adduct 3a is generated and undergoes an exchange with triflate to regenerate the complex A for the next catalytic cycle through the release of product 3a (Scheme 4). Similarly, benzaldehyde (1a) can coordinate with the central metal Y from the bottom side, as well. In this case, the enolic form of acetone attacks the coordinated benzaldehyde (1a) from its bottom side, affording adduct 3a through a similar six-membered ring transition state under the backbone of the tridentate ligand. For the both top and bottom coordination directions of benzaldehyde (1a), it should be located on the left side due to the steric hindrance of the isopropyl group on the right side and favorable π-stacking interaction with the benzene ring in the chiral ligand backbone. The π-stacking interaction is verified by the fact that both aliphatic cyclohexanecarbaldehyde and non-electron-deficient benzaldehydes did not take place in the catalytic asymmetric aldol condensation due to the absence of or weak π-stacking interaction. Because the benzene ring in the chiral ligand backbone is an electron-rich aromatic ring due to its strong electron-donating hydroxyl group, it would exert strong interaction with electron-deficient aromatic aldehydes, forming favorable transition states in the catalytic cycle.
The π-stacking interaction exerts a crucial influence in promoting organic reactions [34] and controlling stereoselectivities in asymmetric organic reactions [35,36,37]. In the current asymmetric reaction, it also plays an important role in controlling the enantioselectivities and diastereoselectivities.
3. Materials and Methods
3.1. Materials and Instruments
Unless otherwise noted, all materials were purchased from commercial suppliers. 1H NMR spectra were recorded on a Bruker 400 NMR spectrometer (Billerica, MA, USA), usually with TMS as an internal standard. Chemical shifts are recorded in ppm relative to tetramethylsilane and with the solvent resonance as the internal standard. Data are reported as follows: chemical shift, multiplicity—singlet (s), doublet (d), triplet (t), quartet (q), double doublet (dd), multiplet (m), and broad (br)—coupling constants (Hz), and integration. 13C NMR data were collected on the same instrument (101 MHz), with complete proton decoupling. Unless otherwise noted, all the solvents were purified using usual methods prior to use. Column chromatography was performed on silica gel (normal phase, 200–300 mesh) from Anhui Liangchen Silicon Material Co., Ltd. (Lu’an, China). Petroleum ether (PE, b.p. 60–90 °C) and ethyl acetate (EA) were used as eluent. Reactions were monitored using thin-layer chromatography (TLC) on GF254 silica gel plates (0.2 mm) from Anhui Liangchen Silicon Material Co., Ltd. The plates were visualized using UV light. Specific rotations were measured on an Anton Paar MCP500 polarimeter (Singapore) and reported as follows: [α]DT (c: g/100 mL, in solvent). The enantiomeric excesses were determined using chiral HPLC analysis using an Agilent 1260 LC instrument (Santa Clara, CA, USA) with Daicel Chiralcel OD-H, AD-H, OJ-H, or AS-H column (Hyderabad, India) with a mixture of isopropyl alcohol and hexane as eluents.
The chiral aziridine-containing vicinal iminophenol tridentate ligands (salazins) were prepared from enantiopure aziridines and salicylaldehydes by referring our previously reported procedure [26].
3.2. General Procedure for the Asymmetric Catalytic Aldol Condensation of Aromatic Aldehydes 1 and Ketones 2
The chiral tridentate salazin ligand (0.02 mmol) and rare-earth metal triflate (0.02 mmol) were stirred in 2 mL of ketone 2 at 40 °C for an hour. After the reaction mixture was cooled to room temperature, aldehyde 1 (0.2 mmol) was added into the mixture. The mixture was stirred at 0 °C for 26 h. The reaction mixture was quenched with 1 mL of saturated aqueous ammonium chloride solution and then extracted with DCM (3 × 5 mL). The combined organic layer was dried over anhydrous Na2SO4. After the evaporation of the solvent under reduced pressure, the crude residue was purified using column chromatography on silica gel using petroleum ether/ethyl acetate (3:1, v/v) as eluent to afford the pure aldol adduct 3 or 5. The enantiomeric excess was determined using chiral HPLC analysis with a mixture of iPrOH and hexane as eluent.
(R)-4-Hydroxy-4-(4-nitrophenyl)butan-2-one (3a). Colorless crystals, 41 mg, yield 98%; m.p. 58–60 °C Lit. [26] m.p. 58–60 °C; = +63.1 (c 0.50, CHCl3), Lit. [26] = +37.0 (c 0.54, CHCl3); Rf = 0.35 (PE/EA = 3:1, v/v), ee 98%, HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 25:75, v/v, 1.0 mL/min, 260 nm) major tR = 17.10 min and minor tR = 24.08 min. 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.8 Hz, 2H), 7.54 (d, J = 8.8 Hz 2H), 5.30–5.24 (m, 1H), 3.68 (d, J = 3.0 Hz, 1H), 2.87 (s, 1H), 2.86 (d, J = 2.7 Hz, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, CHCl3) δ 208.5, 150.0, 147.23, 126.4, 123.7, 68.8, 51.5, 30.7.
(R)-4-Hydroxy-4-(3-nitrophenyl)butan-2-one (3b). Colorless oil, 30 mg, yield 72%, = +65.8 (c 0.54, CHCl3), Lit. [26] = +65.8 (c 0.60, CHCl3); Rf = 0.33 (PE/EA = 3:1, v/v), ee 90%. HPLC analysis: chiralpak AD-H (i-PrOH/hexane = 10:90, v/v, 1.0 mL/min, 260 nm) major tR = 14.76 min and minor tR = 15.51 min. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 8.13 (d, J = 8.2 Hz, 1H), 7.72 (d, J = 7.7 Hz, 1H), 7.53 (t, J = 7.9 Hz, 1H), 5.30–5.23 (m, 1H), 3.72 (d, J = 3.1 Hz, 1H), 2.90 (s, 1H), 2.89 (s, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 208.5, 148.3, 144.9, 131.8, 129.4, 122.5, 120.7, 68.7, 51.5, 30.7.
(R)-4-Hydroxy-4-(2-nitrophenyl)butan-2-one (3c). Yellow oil, 35 mg, yield 84%; = −157.6 (c 0.30, CHCl3), Lit. [26] = −142.6 (c 0.78, CHCl3); Rf = 0.31 (PE/EA = 3:1, v/v), ee 93%. HPLC analysis: chiralpak AD-H (i-PrOH/hexane = 5:95, v/v, 1.0 mL/min, 260 nm) major tR = 22.13 min and minor tR = 23.18 min. 1H NMR (400 MHz, CDCl3) δ 7.96 (dd, J = 8.2, 1.3 Hz, 1H), 7.90 (dd, J = 8.0, 1.4 Hz, 1H), 7.67 (td, J = 7.6, 1.3 Hz, 1H), 7.44 (ddd, J = 8.5, 7.4, 1.5 Hz, 1H), 5.68 (dt, J = 9.5, 2.4 Hz, 1H), 3.76 (d, J = 3.0 Hz, 1H), 3.13 (dd, J = 17.8, 2.1 Hz, 1H), 2.73 (dd, J = 17.8, 9.4 Hz, 1H), 2.24 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 208.8, 147.1, 138.4, 133.8, 128.3, 128.2, 124.4, 65.6, 51.0, 30.4.
(R)-4-Hydroxy-4-(4-cyanophenyl)butan-2-one (3d). Yellow oil, 35 mg, yield 93%; = +74.3 (c 0.48, CHCl3), Lit. [26] = +81.1 (c 0.62, CHCl3); Rf = 0.33 (PE/EA = 3:1, v/v), ee 91%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 30:70, v/v, 1.0 mL/min, 214 nm) major tR = 12.17 min and minor tR = 22.29 min. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.2 Hz, 2H), 7.48 (d, J = 8.3 Hz, 2H), 5.21 (td, J = 6.2, 3.2 Hz, 1H), 3.65 (dt, J = 3.4, 1.6 Hz, 1H), 2.84 (d, J = 6.2 Hz, 2H), 2.21 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 208.4, 148.0, 132.3, 126.3, 118.7, 111.2, 69.0, 51.5, 30.7.
(R)-4-(4-Chlorophenyl)-4-hydroxybutan-2-one (3e). Colorless crystals, 13 mg, yield 33%; m.p. 52–54 °C, Lit. [26] m.p. 52–54 °C; = +70.5 (c 0.50, CHCl3), Lit. [26] = +62.0 (c 0.48, CHCl3); Rf = 0.33 (PE/EA = 3:1, v/v), ee 91%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 15:85, v/v, 1.0 mL/min, 214 nm) major tR = 11.36 min and minor tR = 14.18 min. 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.7 Hz, 2H), 7.26 (d, J = 8.7 Hz, 2H), 5.09 (dt, J = 9.1, 3.0 Hz, 1H), 3.64–3.59 (m, 1H), 2.83 (dd, J = 17.5, 8.9 Hz, 1H), 2.75 (dd, J = 17.5, 3.6 Hz, 1H), 2.17 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 208.7, 141.3, 133.1, 128.5, 126.9, 69.0, 51.7, 30.6.
(R)-4-(4-Bromophenyl)-4-hydroxybutan-2-one (3f). Colorless crystals, 19 mg, yield 39%; m.p. 57–59 °C, Lit. [26] m.p. 57–59 °C; = +54.5 (c 0.51, CHCl3), Lit. [26] = +42.2 (c 0.46, CHCl3), Rf = 0.40 (PE/EA = 3:1, v/v), ee 88%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 20:80, v/v, 1.0 mL/min, 214 nm) major tR = 9.33 min and minor tR = 11.66 min. 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 5.09 (dq, J = 8.8, 2.8 Hz, 1H), 3.51–3.45 (m, 1H), 2.87–2.74 (m, 2H), 2.18 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 208.7, 141.3, 133.1, 128.5, 126.9, 69.0, 51.7, 30.6.
(R)-4-Hydroxy-4-(naphthalen-1-yl)butan-2-one (3j). Colorless crystals, 15 mg, yield 35%; m.p. 99–101 °C; [α]24D = +63.0 (c 0.54, CHCl3), Lit. [38] = +74.3 (c 0.48, CHCl3), Rf = 0.30 (PE/EA = 3:1, v/v), ee 96%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 15:85, v/v, 1.0 mL/min, 210 nm) major tR = 16.91 min and minor tR = 16.22 min. 1H NMR (400 MHz, CDCl3) δ 8.00–7.96 (m, 1H), 7.87–7.83 (m, 1H), 7.76 (dt, J = 8.3, 1.1 Hz, 1H), 7.66 (dt, J = 7.1, 1.0 Hz, 1H), 7.52–7.42 (m, J = 11.6, 8.2, 7.0, 3.3 Hz, 3H), 5.92 (td, J = 6.1, 2.9 Hz, 1H), 3.48 (d, J = 3.3 Hz, 1H), 2.95 (d, J = 6.1 Hz, 2H), 2.19 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 209.1, 138.2, 133.7, 129.8, 128.9, 128.0, 126.1, 125.5, 122.9, 122.7, 66.6, 51.3, 30.7.
(R)-4-Hydroxy-4-(naphthalen-2-yl)butan-2-one (3k). Yellow oil, 36 mg, yield 84%, = +44.7 (c 0.40, CHCl3), Rf = 0.60 (PE/EA = 2:1, v/v), ee 91%, Lit. [26] = +39.8 (c 0.45, CHCl3). HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 15:85, v/v, 1.0 mL/min, 210 nm) major tR = 15.00 min and minor tR = 16.91 min. 1H NMR (400 MHz, CDCl3) δ 7.83–7.72 (m, 4H), 7.47–7.37 (m, 3H), 5.25 (dd, J = 9.2, 3.3 Hz, 1H), 3.61 (br s, 1H), 2.89 (dd, J = 17.4, 9.2 Hz, 1H), 2.79 (dd, J = 17.3, 3.4 Hz, 1H), 2.12 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 208.9, 140.1, 133.1, 132.8, 128.2, 127.9, 127.5, 126.1, 125.8, 124.2, 123.7, 69.8, 51.8, 30.6.
(R)-4-Hydroxy-4-(quinolin-4-yl)butan-2-one (3l). Colorless crystals, 38 mg, yield 89%; m.p. 62–64 °C, = +168.3 (c 0.75, CHCl3), Lit. [39] [α]20D = +37.2 (c 0.1, CHCl3); Rf = 0.20 (PE/EA = 1:1, v/v), ee 89%. HPLC analysis: chiralpak OD-H (i-PrOH/hexane = 20:80, v/v, 1.0 mL/min, 260 nm) minor tR = 10.08 min and major tR = 10.80 min. 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 4.5 Hz, 1H), 8.08 (d, J = 7.9 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 7.66 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.62 (dd, J = 4.5, 0.8 Hz, 1H), 7.52 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 5.97 (t, J = 6.0 Hz, 1H), 4.71 (br s, 1H), 2.92 (d, J = 6.0 Hz, 2H), 2.23 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 207.9, 150.2, 148.8, 147.8, 130.1, 129.1, 126.7, 124.9, 122.5, 117.5, 65.4, 51.1, 30.8.
(R)-4-Hydroxy-4-(4-(methylsulfonyl)phenyl)butan-2-one (3m). Colorless crystals, 41 mg, yield 86%, m.p. 113–115 °C, = +57.2 (c 0.37, CHCl3), Rf = 0.15 (PE/EA = 1:1, v/v), ee 92%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 40:60, v/v, 1.0 mL/min, 214 nm) major tR = 22.36 min and minor tR = 35.43 min. 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.3 Hz, 2H), 5.24 (dd, J = 7.4, 5.0 Hz, 1H), 3.81 (s, 1H), 3.04 (s, 3H), 2.86 (d, J = 2.9 Hz, 1H), 2.85 (s, 1H), 2.21 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 208.4, 149.1, 139.4, 127.5, 126.5, 68.9, 51.5, 44.4, 30.6. HRMS (ESI): m/z calcd for C11H14NaO4S+ [M+Na]+: 265.0505, found: 265.0513.
(E)-4-(4-(Methylsulfonyl)phenyl)but-3-en-2-one (4m). Colorless crystals, 6 mg, yield 14%, m.p. 125–126 °C, Lit. [40] m.p. 124–125 °C. Rf = 0.40 (PE/EA = 3:1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.00–7.95 (m, 2H), 7.76–7.71 (m, 2H), 7.53 (d, J = 16.3 Hz, 1H), 6.82 (d, J = 16.3 Hz, 1H), 3.08 (s, 3H), 2.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 197.6, 141.6, 140.5, 139.8, 130.0, 128.8, 128.1, 44.4, 28.0.
(R)-4-Hydroxy-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)butan-2-one (3n). Colorless crystals, 41 mg, yield 70%, m.p. 58–60 °C, = +52.2 (c 0.37, CHCl3), Rf = 0.40 (PE/EA = 3:1, v/v), ee 87%. HPLC analysis: chiralpak AD-H (i-PrOH/hexane = 10:90, v/v, 1.0 mL/min, 230 nm) major tR = 7.18 min and minor tR = 8.24 min. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 5.16 (dt, J = 8.9, 3.1 Hz, 1H), 3.36 (d, J = 3.1 Hz, 1H), 2.86 (dd, J = 17.5, 8.9 Hz, 1H), 2.79 (dd, J = 17.5, 3.5 Hz, 1H), 2.18 (s, 3H), 1.34 (s, 12H). 13C NMR (101 MHz, CDCl3) δ 208.9, 145.8, 135.0 (2C), 124.8, 83.8, 69.8, 51.9, 30.7, 24.8. HRMS (ESI): m/z calcd for C16H23BNaO4+ [M+Na]+: 313.1582, found: 313.1585.
(S/R)-2-((R/S)-Hydroxy(4-nitrophenyl)methyl)cyclobutan-1-one (5a). Mixture of anti- and syn-diastereomers, yellow solid, 11 mg, yield 25%; m.p. 98–101 °C, Lit. [41] m.p. of syn-diastereomer 101–103 °C, m.p. of anti-diastereomer 97–99 °C; = +19.0 (c 0.27, CHCl3); Rf = 0.25 (PE/EA =3:1, v/v), dr anti:syn = 53:47, ee of anti 83%, ee of syn 69%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 10:90, v/v, 1.0 mL/min, 260 nm) syn-diastereomer: minor tR = 20.96 and major tR = 24.21 min. anti-diastereomer: major tR = 28.10 min and minor tR = 52.43 min. 1H NMR (400 MHz, CDCl3) δ 8.29–8.18 (m, 2H), 7.61–7.48 (m, 2H), 5.34–4.94 (m, 1H), 3.67–3.57 (m, 1H), 3.19–3.08 (m, 1H), 3.04–2.95 (m, 1H), 2.88–2.84 (m, 1H), 2.26–2.06 (m, 1H), 1.99–1.88 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 209.2, 208.5, 149.9, 148.3, 127.0, 126.4, 123.8, 123.8, 73.0, 68.9, 66.0, 51.5, 45.2, 30.7, 14.1.
(S)-2-(Hydroxy(4-nitrophenyl)methyl)cyclopentan-1-one (5b). Mixture of anti- and syn-diastereomers, yellow crystals, 38 mg, yield 81%; m.p. 89–91 °C, Lit. [42] m.p. 89–91 °C; = +124.0 (c 0.38, CHCl3), Lit. [43] = +98 (c 1.5, CHCl3); Rf = 0.28 (PE/EA = 3:1, v/v), dr anti:syn = 83:17, ee of anti 96%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 10:90, v/v, 1.0 mL/min, 210 nm) syn-diastereomer: tR = 36.39 min (enantiomers are inseparable); anti-diastereomer minor tR = 41.78 min and major tR = 59.270 min. 1H NMR (400 MHz, CDCl3) δ 8.23–8.18 (m, 2H), 7.54 (d, J = 8.4 Hz, 2H, anti), 7.54 (d, J = 9.2 Hz, 2H, syn), 5.42 (d, J = 2.8 Hz, 1H, syn), 4.86 (d, J = 9.2 Hz, 1H, anti), 4.77 (br s, 1H, anti), 4.38–4.33 (m, 1H, syn), 2.48–2.35 (m, 2H), 2.38–2.17 (m, 1H), 2.07–1.97 (m, 1H), 1.73–1.68 (m, 2H), 1.58–1.48 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 222.2, 148.6, 147.7, 127.3, 123.6, 74.3, 55.0, 38.5, 26.8, 20.3.
(S)-2-((R)-Hydroxy(4-nitrophenyl)methyl)cyclohexan-1-one (5c). Colorless crystals, 47 mg, yield 94%; m.p. 129–130 °C, Lit. [42] m.p. 129–130 °C; = +11.1 (c 0.17, CHCl3), Lit. [42] = +12.8 (c 1.85, CHCl3); Rf = 0.30 (PE/EA = 3:1, v/v), dr anti:syn = 94:6, ee of anti 91%, ee of syn 5%. HPLC analysis: chiralpak AS-H (i-PrOH/hexane = 15:85, v/v, 1.0 mL/min, 210 nm) syn-diastereomer: major tR = 14.62 min and minor tR = 15.57 min; anti-diastereomer: minor tR = 16.92 min and major tR = 22.93 min. 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.7 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 4.91 (dd, J = 8.3, 2.2 Hz, 1H), 4.11 (d, J = 3.1 Hz, 1H), 2.67–2.58 (m, 1H), 2.53–2.46 (m, 1H), 2.37 (td, 13.2, 6.0 Hz, 1H), 2.16–2.08 (m, 1H), 1.87–1.79 (m, 1H), 1.73–1.65 (m, 1H), 1.63–1.52 (m, 2H), 1.40 (td, J = 13.2, 4.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 214.7, 148.4, 147.5, 127.8, 123.5, 73.9, 57.1, 42.6, 30.7, 27.6, 24.6.
(1R,2S)-1-Hydroxy-2-methyl-1-(4-nitrophenyl)pentan-3-one (5d). Colorless oil, 14 mg, yield 30%; = +27.3 (c 0.08, CHCl3), Lit. [43] = +26 (c 1.0, CHCl3),Rf = 0.53 (PE/EA = 3:1, v/v), dr anti:syn = 86:14, ee of anti 98%, ee of syn 71%. HPLC analysis: chiralpak OJ-H (i-PrOH/hexane = 10:90, v/v, 1.0 mL/min, 260 nm) syn-diastereomer major tR = 14.92 min and minor tR = 16.25 min; anti-diastereomer major tR = 13.44 min and minor tR = 17.29 min. 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.6 Hz, 2H), 5.32–4.81 (m, 1H), 3.58 (br s, 1H), 2.99–2.79 (m, 1H), 2.61 (dq, J = 18.2, 7.2 Hz, 1H), 2.46 (dq, J = 18.1, 7.2 Hz, 1H), 1.07 (d, J = 7.2 Hz, 3H), 1.04 (d, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 216.2, 149.1, 127.3, 126.8, 124.3, 72.0, 51.4, 35.1, 9.9, 7.5.
(R)-3-Hydroxy-3-(4-nitrophenyl)-1-phenylpropan-1-one (5e). Colorless crystals, 8 mg, yield 15%; m.p. 112–113 °C, Lit. [44] m.p. 113–114 °C; [α]24D = +52.3 (c 1.0, CHCl3), Lit. [45] = +13.7 (c 0.06, CHCl3), Rf = 0.52 (PE/EA = 3:1, v/v), ee 44%. HPLC analysis: chiralpak AD-H (i-PrOH/hexane = 10:90, v/v, 1.0 mL/min, 254 nm), minor tR = 31.19 min and major tR = 39.00 min. 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.8 Hz, 2H), 7.94 (d, J = 7.1 Hz, 2H), 7.61 (d, J = 8.7 Hz, 3H), 7.48 (t, J = 7.8 Hz, 2H), 5.46 (dt, J = 8.4, 3.4 Hz, 1H), 3.91 (d, J = 3.1 Hz, 1H), 3.41 (dd, J = 17.9, 3.6 Hz, 1H), 3.35 (dd, J = 17.9, 8.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 199.4, 150.2, 147.3, 136.1, 134.0, 128.8, 128.1, 126.5, 123.7, 69.1, 46.9.
4. Conclusions
The chiral aziridine-containing vicinal iminophenol tridentate ligands (salazins) are a class of easily synthesized chiral ligands from readily available prepared enantiopure aziridines and salicylaldehydes. Both of their scandium and yttrium triflate complexes show excellent enantioselectivity in the catalytic asymmetric aldol condensation of electron-deficient aromatic aldehydes and ketones and both excellent diastereo- and enantioselectivity in the reactions with cycloalkanones. The stereoselectivities are attributed to the strong π–stacking interaction between aromatic aldehydes and the vicinal iminophenol group in the chiral ligands.
Supplementary Materials
The following supporting information can be downloaded at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/molecules29091963/s1. Copies of 1H-NMR and 13C-NMR spectra, HPLC profiles of compounds 3 and 5, and HRMS spectra of unknown products 3 are included in the Supplementary Materials.
Author Contributions
Conceptualization, J.X.; methodology, C.W.; validation, C.W., Z.Y., N.C. and J.X.; formal analysis, N.C.; investigation, C.W.; data curation, N.C.; writing—original draft preparation, J.X.; writing—review and editing, J.X. and Z.Y.; visualization, J.X.; supervision, J.X.; project administration, J.X.; funding acquisition, J.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key R&D Program of China (No. 2022YFF0709803).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Schmidt, F.; Keller, F.; Vedrenne, E.; Aggarwal, V.K. Stereocontrolled Synthesis of β-Amino Alcohols from Lithiated Aziridines and Boronic Esters. Angew. Chem. Int. Ed. Engl. 2009, 48, 1149–1152. [Google Scholar] [CrossRef]
- Tanner, D. Stereocontrolled synthesis via chiral aziridines. Pure Appl. Chem. 1993, 65, 1319–1328. [Google Scholar] [CrossRef]
- Zhu, M.; Hu, L.B.; Chen, N.; Du, D.-M.; Xu, J.X. Synthesis of NH-aziridines from vicinal amino alcohols via the Wenker reaction: Scope and limitation. Lett. Org. Chem. 2008, 5, 212–217. [Google Scholar] [CrossRef]
- Li, X.Y.; Chen, N.; Xu, J.X. An improved and mild Wenker synthesis of aziridines. Synthesis 2010, 2010, 3423–3428. [Google Scholar]
- Jarzyński, S.; Utecht, G.; Leśniak, S.; Rachwalski, M. Highly enantioselective asymmetric reactions involving zinc ions promoted by chiral aziridine alcohols. Tetrahedron Asymmetry 2017, 28, 1774–1779. [Google Scholar] [CrossRef]
- Jarzyński, S.; Leśniak, S.; Pieczonka, A.M.; Rachwalski, M. N-Trityl-aziridinyl alcohols as highly efficient chiral catalysts in asymmetric additions of organozinc species to aldehydes. Tetrahedron Asymmetry 2015, 26, 35–40. [Google Scholar] [CrossRef]
- Rachwalski, M.; Jarzyński, S.; Jasiński, M.; Leśniak, S. Mandelic acid derived α-aziridinyl alcohols as highly efficient ligands for asymmetric additions of zinc organyls to aldehydes. Tetrahedron Asymmetry 2013, 24, 689–693. [Google Scholar] [CrossRef]
- Pieczonka, A.M.; Leśniak, S.; Jarzyński, S.; Rachwalski, M. Aziridinylethers as highly enantioselective ligands for the asymmetric addition of organozinc species to carbonyl compounds. Tetrahedron Asymmetry 2015, 26, 148–151. [Google Scholar] [CrossRef]
- Pieczonka, A.M.; Jarzyński, S.; Wujkowska, Z.; Leśniak, S.; Rachwalski, M. Zinc(II) mediated asymmetric aldol condensation catalyzed by chiral aziridine ligands. Tetrahedron Lett. 2015, 56, 6506–6507. [Google Scholar] [CrossRef]
- Jarzyński, S.; Rachwalski, M.; Pieczonka, A.M.; Wujkowska, Z.; Leśniak, S. Highly efficient conjugate additions of diethylzinc to enones promoted by chiral aziridine alcohols and aziridine ethers. Tetrahedron Asymmetry 2015, 26, 924–927. [Google Scholar] [CrossRef]
- Rachwalski, M.; Jarzyński, S.; Leśniak, S. Aziridine ring-containing chiral ligands as highly efficient catalysts in asymmetric synthesis. Tetrahedron Asymmetry 2013, 24, 421–425. [Google Scholar] [CrossRef]
- Adam, E.M.; Pieczonka, M.; Rachwalski, M.; Leśniak, S. Synthesis of chiral 1-(2-aminoalkyl)aziridines via the self-opening reaction of aziridine. ARKIVOC 2017, 2017, 223–234. [Google Scholar]
- Pieczonka, A.M.; Marciniak, L.; Rachwalski, M.; Leśniak, S. Enantiodivergent aldol condensation in the presence of aziridine/acid/water systems. Symmetry 2020, 12, 930. [Google Scholar] [CrossRef]
- Pieczonka, A.M.; Leśniak, S.; Rachwalski, M. Direct asymmetric aldol condensation catalyzed by aziridine semicarbazide zinc(II) complexes. Tetrahedron Lett. 2014, 55, 2373–2375. [Google Scholar] [CrossRef]
- Leśniak, S.; Pieczonka, A.M.; Jarzyński, S.; Justyna, K.; Rachwalski, M. Synthesis and evaluation of the catalytic properties of semicarbazides derived from N-triphenylmethyl-aziridine-2-carbohydrazides. Tetrahedron Asymmetry 2013, 24, 1341–1344. [Google Scholar] [CrossRef]
- Buchcic-Szychowska, A.; Zawisza, A.; Le’sniak, S.; Rachwalski, M. Highly efficient asymmetric Morita–Baylis–Hillman reaction promoted by chiral aziridine-phosphines. Catalysts 2022, 12, 394. [Google Scholar] [CrossRef]
- Buchcic-Szychowska, A.; Lésniak, S.; Rachwalski, M. Chiral aziridine phosphines as highly effective promoters of asymmetric Rauhut–Currier reaction. Symmetry 2022, 14, 1631. [Google Scholar] [CrossRef]
- Buchcic, A.; Zawisza, A.; Lésniak, S.; Adamczyk, J.; Pieczonka, A.M.; Rachwalski, M. Enantioselective Mannich reaction promoted by chiral phosphinoyl-aziridines. Catalysts 2019, 9, 837. [Google Scholar] [CrossRef]
- Tanner, D.; Johansson, F.; Harden, A.; Andersson, P.G. A comparative study of C2-symmetric bis(aziridine) ligands in some transition metal-mediated asymmetric transformations. Tetrahedron 1998, 54, 15731–15738. [Google Scholar] [CrossRef]
- Tanner, D.; Harden, A.; Johansson, F.; Wyatt, P.; Andersson, P.G.; Zhang, S.Y.; Zhao, S.H.; Ciglic, M.I.; Haugg, M.; Trabesinger-Rüf, N.; et al. Asymmetric catalysis via chiral aziridines. Acta Chem. Scand. 1996, 50, 361–368. [Google Scholar] [CrossRef]
- Andersson, P.G.; Harden, A.; Tanner, D.; Norrby, P.-O. Studies of Allylic Substitution Catalysed by a Palladium Complex of a C2-Symmetric Bis(aziridine): Preparation and NMR Spectroscopic Investigation of a Chiral π-Allyl Species. Chem.-Eur. J. 1995, 1, 12–16. [Google Scholar] [CrossRef]
- Tanner, D.; Andersson, P.G.; Harden, A.; Somfai, P. C2-symmetric bis(aziridines): A new class of chiral ligands for transition metal-mediated asymmetric synthesis. Tetrahedron Lett. 1994, 35, 4631–4634. [Google Scholar] [CrossRef]
- Rachwalski, M.; Leśniak, S.; Kiełbasiński, P. Highly enantioselective addition of phenylethynylzinc to aldehydes using aziridine-functionalized tridentate sulfinyl ligands. Tetrahedron Asymmetry 2010, 21, 2687–2689. [Google Scholar] [CrossRef]
- Rachwalski, M.; Leśniak, S.; Kiełbasiński, P. Highly enantioselective conjugate addition of diethylzinc to enones using aziridine-functionalized tridentate sulfinyl ligands. Tetrahedron Asymmetry 2010, 21, 1890–1892. [Google Scholar] [CrossRef]
- Leśniak, S.; Rachwalski, M.; Sznajder, E.; Kiełbasiński, P. New highly efficient aziridine-functionalized tridentate sulfinyl catalysts for enantioselective diethylzinc addition to carbonyl compounds. Tetrahedron Asymmetry 2009, 20, 2311–2314. [Google Scholar] [CrossRef]
- Chen, X.P.; Lin, C.; Du, H.G.; Xu, J.X. Efficient direct synthesis of aziridine-containing chiral tridentate ligands by the iminium-mediated self-ring opening reaction of enantiopure aziridines and salicylaldehydes. Adv. Synth. Catal. 2019, 361, 1647–1661. [Google Scholar] [CrossRef]
- Ma, L.G.; Xu, J.X. Nucleophilic ring opening reaction of unsymmetric aziridines and its regioselectivity. Prog. Chem. (Huaxue Jinzhan) 2004, 16, 220–235. [Google Scholar]
- Wu, Y.-H.; Zhang, L.-Y.; Wang, N.-X.; Xing, Y.L. Recent advances in the rare-earth metal triflates catalyzed organic reactions. Catal. Rev. 2020, 64, 679–715. [Google Scholar] [CrossRef]
- Feng, X.M.; Wang, Z.; Liu, X.L. Chiral Lewis acid rare-earth metal complexes in enantioselective catalysis. In Topics in Organometallic Chemistry; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Ma, L.G.; Jiao, P.; Zhang, Q.H.; Du, D.-M.; Xu, J.X. Ligand and substrate π-stacking interaction contro-lled enantioselectivity in the asymmetric aziridination. Tetrahedron Asymmetry 2007, 18, 878–884. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Chen, Y.; Zheng, C.-Q.; Yin, Y.; Wang, L.; Sun, X.-Q. Highly enantioselective aldol reactions catalyzed by reusable upper rim-functionalized calix[4]arene-based l-proline organocatalyst in aqueous conditions. Tetrahedron 2017, 73, 78–85. [Google Scholar] [CrossRef]
- Vlasserou, I.; Sfetsa, M.; Gerokonstantis, D.-T.; Kokotos, C.G.; Moutevelis-Minakakis, P. Combining prolinamides with 2-pyrrolidinone: Novel organocatalysts for the asymmetric aldol reaction. Tetrahedron 2018, 74, 2338–2349. [Google Scholar] [CrossRef]
- Chen, G.; Fu, X.; Li, C.; Wu, C.; Miao, Q. Highly efficient direct a larger-scale aldol reactions catalyzed by a flexible prolinamide based-metal Lewis acid bifunctional catalyst in the presence of water. J. Organomet. Chem. 2012, 702, 19–26. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Chen, Y.X.; Xu, J.X. π-Stacking-controlled dearomatic sulfur-shifted ene reaction of ketenes and polycyclic arylthiiranes: Access to areno[d]-ε-thiolactones. J. Org. Chem. 2024, 89, 4749–4759. [Google Scholar] [CrossRef] [PubMed]
- Li, B.N.; Wang, Y.K.; Du, D.-M.; Xu, J.X. Notable and obvious ketene substituent-dependent effect of temperature on the stereoselectivity in the Staudinger reaction. J. Org. Chem. 2007, 72, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.X. Recent advances in π-stacking interaction-controlled asymmetric synthesis. Molecules 2024, 29, 1454. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Yu, M.; Wu, X.; Zhang, Y.; Zhao, G. 4,4′-Disubstituted L-prolines as highly enantioselective catalysts for direct aldol reactions. Adv. Synth. Catal. 2006, 348, 2223–2228. [Google Scholar] [CrossRef]
- Tang, Z.; Jiang, F.; Yu, L.T.; Cui, X.; Gong, L.Z.; Mi, A.Q.; Jiang, Y.Z.; Wu, Y.D. Novel small organic molecules for a highly enantioselective direct aldol reaction. J. Am. Chem. Soc. 2003, 125, 5262–5263. [Google Scholar] [CrossRef] [PubMed]
- Kucherenko, A.S.; Kostenko, A.A.; Gerasimchuk, V.V.; Zlotin, S.G. Stereospecific diaza-Cope rearrangement as an efficient tool for the synthesis of DPEDA pyridine analogs and related C2-symmetric organocatalysts. Org. Biomol. Chem. 2017, 15, 7028–7033. [Google Scholar] [CrossRef]
- Edwards, M.L.; Ritter, H.W.; Stemerick, D.M.; Stewart, K.T. Mannich bases of 4-phenyl-3-buten-2-one. A new class of antiherpes agent. J. Med. Chem. 1983, 26, 431–436. [Google Scholar] [CrossRef]
- Cobb, A.J.A.; Shaw, D.M.; Longbottom, D.A.; Gold, J.B.; Ley, S.V. Organocatalysis with proline derivatives: Improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org. Biomol. Chem. 2005, 3, 84–96. [Google Scholar] [CrossRef]
- Guo, G.; Wu, Y.; Zhao, X.; Wang, J.; Zhang, L.; Cui, Y. Polymerization of L-proline functionalized styrene and its catalytic performance as a supported organocatalyst for direct enantioselective aldol reaction. Tetrahedron Asymmetry 2016, 27, 740–746. [Google Scholar] [CrossRef]
- Da, C.-S.; Che, L.-P.; Guo, Q.-P.; Wu, F.-C.; Ma, X.; Jia, Y.-N. 2,4-Dinitrophenol as an effective cocatalyst: Greatly improving the activities and enantioselectivities of primary amine organocatalysts for asymmetric aldol reactions. J. Org. Chem. 2009, 74, 2541–2546. [Google Scholar] [CrossRef] [PubMed]
- Downey, C.W.; Johnson, M.W. A tandem enol silane formation-Mukaiyama aldol reaction mediated by TMSOTf. Tetrahedron Lett. 2007, 48, 3559–3562. [Google Scholar] [CrossRef]
- Li, L.; Gou, S.H.; Liu, F. Highly stereoselective direct aldol reactions catalyzed by a bifunctional chiral diamine. Tetrahedron Asymmetry 2014, 25, 193–197. [Google Scholar] [CrossRef]
Scheme 1.
Aziridine-containing chiral ligands and application of chiral aziridine-containing vicinal iminophenol tridentate ligands in the catalytic asymmetric aldol condensation.
Scheme 1.
Aziridine-containing chiral ligands and application of chiral aziridine-containing vicinal iminophenol tridentate ligands in the catalytic asymmetric aldol condensation.

Scheme 2.
Catalytic asymmetric aldol condensation under the catalysis of Y(OTf)3 a. a Reactions were conducted on a 0.2 mmol scale of aldehyde in 2 mL of acetone. The yields were obtained using chromatography on silica gel. b Dehydrated side product 4-(4-methanesulfonylphenyl)but-3-en-2-one (4m) was obtained in a 14% yield. c Reaction was conducted for 70 h.
Scheme 2.
Catalytic asymmetric aldol condensation under the catalysis of Y(OTf)3 a. a Reactions were conducted on a 0.2 mmol scale of aldehyde in 2 mL of acetone. The yields were obtained using chromatography on silica gel. b Dehydrated side product 4-(4-methanesulfonylphenyl)but-3-en-2-one (4m) was obtained in a 14% yield. c Reaction was conducted for 70 h.

Scheme 3.
Catalytic asymmetric aldol condensations of 4-benzaldehyde with pentan-3-one and acetophenone.
Scheme 3.
Catalytic asymmetric aldol condensations of 4-benzaldehyde with pentan-3-one and acetophenone.

Scheme 4.
Proposed mechanism for the asymmetric catalytic aldol condensation of benzaldehyde and acetone under the catalysis of Y(OTf)3-tridentate ligand complex. ‡ Indicates that the structure is transition state.
Scheme 4.
Proposed mechanism for the asymmetric catalytic aldol condensation of benzaldehyde and acetone under the catalysis of Y(OTf)3-tridentate ligand complex. ‡ Indicates that the structure is transition state.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Catalytic asymmetric aldol condensation in different solvents a.
 | |||
|---|---|---|---|
| Entry | Solvent | Yield/% | ee/% |
| 1 | PhMe | 3 | 38 |
| 2 | EtOH | 98 | 53 |
| 3 | DCM | 10 | 60 |
| 4 | THF | 20 | 49 |
| 5 | acetone | 96 | 87 |
a All the reactions were carried out with 4-nitrobenzaldehyde (31 mg, 0.2 mmol, 1 eq.) and acetone (12 mg, 0.2 mmol, 1 eq.) in 2 mL of solvent in a 10 mL reaction tube under the catalysis of chiral ligand salazin-Bn (7.4 mg, 0.02 mmol, 0.1 eq.) and Sc(OTf)3 (9.8 mg, 0.02 mol, 0.1 eq.) under stirring for 40 h. The yields were obtained using chromatography on silica gel.
Table 2.
Catalytic asymmetric aldol condensation under the catalysis of different ligands and rare-earth metal triflates at different reaction temperatures a.
Table 2.
Catalytic asymmetric aldol condensation under the catalysis of different ligands and rare-earth metal triflates at different reaction temperatures a.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Metal Salt | Ligand | Temp./°C | Time/h | Yield/% | ee/% |
| 1 | Sc(OTf)3 | Salazin-Bn | 15 | 40 | 96 | 81 |
| 2 | Sc(OTf)3 | Salazin-tBu-Bn | 15 | 40 | 96 | 70 |
| 3 | Sc(OTf)3 | Salazin-Br-Bn | 15 | 40 | 55 | 78 |
| 4 | Sc(OTf)3 | Salazin-NO2-iPr | 15 | 40 | 91 | 86 |
| 5 | Sc(OTf)3 | Salazin-Br-iPr | 15 | 40 | 89 | 87 |
| 6 | Sc(OTf)3 | Salazin-I-iPr | 15 | 40 | 60 | 80 |
| 7 | Sc(OTf)3 | Salazin-iPr | 15 | 26 | 98 | 88 |
| 8 | Y(OTf)3 | Salazin-iPr | 15 | 26 | 98 | 88 |
| 9 | La(OTf)3 | Salazin-iPr | 15 | 26 | 93 | 83 |
| 10 | Ce(OTf)3 | Salazin-iPr | 15 | 26 | 91 | 86 |
| 11 | Eu(OTf)3 | Salazin-iPr | 15 | 26 | 98 | 85 |
| 12 | Gd(OTf)3 | Salazin-iPr | 15 | 26 | 98 | 87 |
| 13 | Lu(OTf)3 | Salazin-iPr | 15 | 26 | 96 | 87 |
| 14 | Zn(OTf)2 | Salazin-iPr | 15 | 40 | 90 | 70 |
| 15 | Sc(OTf)3 | Salazin-iPr | 40 | 20 | 98 | 85 |
| 16 | Sc(OTf)3 | Salazin-iPr | 20 | 26 | 98 | 88 |
| 17 | Sc(OTf)3 | Salazin-iPr | 0 | 30 | 98 | 98 |
| 18 | Y(OTf)3 | Salazin-iPr | 40 | 20 | 96 | 89 |
| 19 | Y(OTf)3 | Salazin-iPr | 20 | 26 | 98 | 88 |
| 20 | Y(OTf)3 | Salazin-iPr | 0 | 30 | 98 | 98 |
a Reactions were conducted on a 0.2 mmol scale of aldehyde in 2 mL of acetone. The yields were obtained using chromatography on silica gel.
Table 3.
Catalytic asymmetric aldol condensation of cycloalkanones under the catalysis of Y(OTf)3 a.
Table 3.
Catalytic asymmetric aldol condensation of cycloalkanones under the catalysis of Y(OTf)3 a.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Product 5 | n | Yield/% | anti:syn | ee of anti/% | ee of syn/% |
| 1 | 5a | 1 | 25 | 53:47 | 83 | 69 |
| 2 | 5b | 2 | 81 | 83:17 | 96 | - b |
| 3 | 5c | 3 | 94 | 94:6 | 91 | 5 |
a Reactions were conducted on a 0.2 mmol scale of 4-nitrobenzaldehyde (1a) in 2 mL of cycloalkanone (2b–2d). The yields were obtained using chromatography on silica gel. b The syn-diastereomer syn-5b cannot be separated in all of our available chiral columns.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, C.; Chen, N.; Yang, Z.; Xu, J. Y(OTf)3-Salazin-Catalyzed Asymmetric Aldol Condensation. Molecules 2024, 29, 1963. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091963
AMA Style
Wang C, Chen N, Yang Z, Xu J. Y(OTf)3-Salazin-Catalyzed Asymmetric Aldol Condensation. Molecules. 2024; 29(9):1963. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091963
Chicago/Turabian StyleWang, Chengzhuo, Ning Chen, Zhanhui Yang, and Jiaxi Xu. 2024. "Y(OTf)3-Salazin-Catalyzed Asymmetric Aldol Condensation" Molecules 29, no. 9: 1963. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091963