
Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds


1
Faculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia
2
Department of Chemistry, College of Science, Sultan Qaboos University, Muscat 123, Oman
*
Author to whom correspondence should be addressed.
Molecules 2024, 29(9), 1917; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091917
Submission received: 28 March 2024
/
Revised: 18 April 2024
/
Accepted: 19 April 2024
/
Published: 23 April 2024
(This article belongs to the Special Issue Feature Papers in Applied Chemistry: 3rd Edition)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Functionalization of C-H bonds has emerged as a powerful strategy for converting inert, nonfunctional C-H bonds into their reactive counterparts. A wide range of C-H bond functionalization reactions has become possible by the catalysis of metals, typically from the second row of transition metals. First-row transition metals can also catalyze C-H functionalization, and they have the merits of greater earth-abundance, lower cost and better environmental friendliness in comparison to their second-row counterparts. C-H bond alkylation is a particularly important C-H functionalization reaction due to its chemical significance and its applications in natural product synthesis. This review covers Ni-catalyzed C-H bond alkylation reactions using alkyl halides and olefins as alkyl sources.
1. Introduction
Functionalization of C-H bonds has emerged as a powerful strategy for converting inert, nonfunctional C-H bonds into their corresponding reactive counterparts [1,2,3,4,5,6,7]. Thus kindly delete the last part of original statement which is highlighted beginning with “C-Metal…..reagents. Since C-H bonds are ubiquitous in organic molecules, controlling which C-H bond to cleave and functionalize is a formidable challenge. The problem of regioselectivity, or site-selectivity, has been partially addressed using monodentate or bidentate directing groups [8,9,10]. Such groups are Lewis bases and can interact with the Lewis acidic metal by bringing it in proximity to a C-H bond to be cleaved. Thus, transition-metal catalyzed, selective activation of C-H bonds, followed by their functionalization to directly form valuable C-C or C–X bonds (X = N, O or S), has revolutionized organic synthesis by enabling the assembly of complex molecular structures from simple precursors. In contrast to conventional cross-coupling reactions (e.g., Suzuki, Negishi or Stille) [11,12,13], C-H bond functionalization does not require the use of pre-functionalized starting materials, such as organometallic reagents, thus reducing the number of synthetic steps and the amount of waste. From this point of view, C-H bond functionalization contributes to more economically and ecologically compatible synthesis protocols, and thus also follows the principles of green chemistry [14]. Successful direct functionalization of C-H bonds was initially achieved using powerful catalysts, typically second-row transition metals such as Pd- [15,16,17,18], Rh- [19,20,21,22,23] or Ru-complexes [24,25,26,27,28], which tolerate a wide range of functionalities and common oxidants and are often stable in air and water [29,30]. Despite their great success in organic synthesis, the low abundance and high price or toxicity of some of these metals necessitated the development of new C-H bond functionalization protocols using non-noble metals. Thus, the strategy was extended to more earth-abundant first-row transition metals [31], such as Ni [32,33,34], Mn [35,36], Fe [37,38] and Co [39,40,41,42,43]. Nickel appears to be an adequate replacement for palladium, as it lies in the same group of the periodic table and can often perform the same reactions as palladium; more importantly, its use in organic synthesis could also lead to the discovery of novel reactions [44]. Various nickel oxidation states, from Ni(0) to Ni(IV), allow for different redox pathways in the catalytic cycle of a given reaction. In fact, nickel catalysts have been widely used for reactions involving alkenes and alkynes, such as oligomerization or cross-couplings [45,46]. While progress in the Ni-catalyzed activation/functionalization of C-H bonds has only recently been advanced to a synthetic level comparable to the direct functionalization of C-H bonds catalyzed by late or noble transition metals, Kleiman and Dubeck reported the formation of a nickelacycle from the stoichiometric reactions of azobenzene and NiCp2 complexes as early as 1963 [47]. However, the catalytic functionalization of C-H bonds by nickel complexes was initially limited to acidic C-H bonds in certain aromatic systems, and there was a lack of general and reliable methods for the activation of non-acidic C-H bonds in various organic substrates, such as azoles [48]. There is no doubt that the alkylation reaction, i.e., the coupling between an alkylating agent and a nucleophile, is an important C-C bond-forming reaction and has been widely applied in organic synthesis. In this review, we focus on the alkylation reactions of C-H bonds mediated by nickel compounds as applied to a variety of organic substrates that can be found in the literature to date, while other nickel-catalyzed C-H bond functionalizations have been reviewed elsewhere [49,50].
There are other modern and significant strategies for C(sp3)–H functionalization. Dearomatization by the hydride transfer strategy has emerged as a powerful method for achieving the ultimate goal of C-H bond functionalization [51]. The hydride transfer strategy has enabled access to various complex heterocycles through a cascade of reactions involving dearomatization and [5+1] and [5+2] cyclizations [52]. In addition, functionalization of C-H bonds has been achieved by the 1,5-hydrogen atom transfer (1,5-HAT) strategy [53].
Although the use of nickel in the direct functionalization of C-H bonds is an attractive area of research, and could certainly compete with other metal-catalyzed reactions in the future, synthetic methods for nickel-catalyzed C-H bond alkylation are rather scarce.
A related review covering Ni-catalyzed C-H bond functionalization reactions was reported [54]. The recent review specifically addressed intramolecular and intermolecular Ni-catalyzed functionalization reactions towards synthesis of N-containing heterocycles in the period 2008–2021 [54].
In this review, emphasis is placed on the alkylation reactions of C-H bonds mediated by nickel compounds as applied to a variety of organic substrates that can be found in the literature to date (2004–2022). The review covers the following two main strategies for transition metal-catalyzed C-H bond alkylation: either (i) using alkenes as alkylating agents by adding the substrate C-H bond across the C=C double bond of the alkene, or (ii) introducing an alkyl moiety by using alkyl halides (or their relatives) as coupling partners, the first strategy being more economical and generating less waste (e.g., no base required) (Scheme 1).
The present review covers methods for Ni-catalyzed C-H bond alkylation reactions to various carbocyclic and heterocyclic substrates with alkyl halides and olefins. In addition, the review coverage includes the use of directed Ni-catalyzed C-H bond alkylation reactions by chelation assistance using directing groups.
2. Discussion
2.1. C-H Bond Alkylation with Alkenes
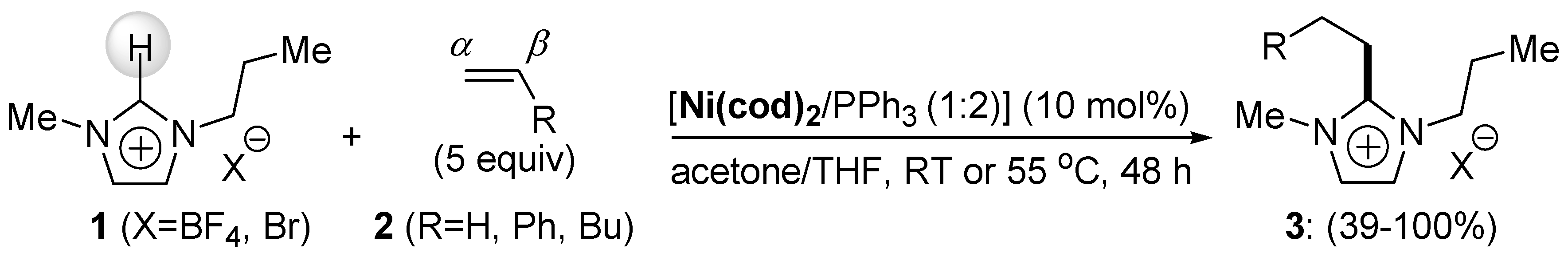
In 2004, Cavell et al. reported a seminal work on Ni(0)-catalyzed C-H bond functionalization in which imidazolium salts 1 were successfully 2-C-alkylated with ethylene and other 1-alkenes 2 in the presence of the Ni(cod)2/PPh3 catalytic system, leading predominantly to linear products 3, in which a C-C bond was formed between the α-carbon of the 1-alkene and the 2-C of substrate 1 (Scheme 2) [55]. The reactivity of 1-alkenes when coupled with imidazolium salts seems to be determined by steric elements, since ethylene (1 bar) was more reactive than 1-hexene. Both ionic liquids, tetrafluoroborate and bromide, were equally reactive with the alkenes used in this study.
Later, the same Ni(0) catalyst precursor, but using PCyp3 as a ligand, was used for the alkylation of pentafluorobenzene with alkenes (2-vinylnaphthalene and (E)-buta-1,3-dien-1-ylbenzene), as shown by Hiyama and coworkers (Scheme 3) [56]. A significant influence of the ligands on the yield of the reaction was observed, as PMe3, PBu3 or PtBu3 ligands were much less effective and gave only trace amounts of coupling products. In contrast to Cavell’s work (vide supra), the alkene in this particular reaction reacted preferentially at the β-carbon atom with pentafluorobenezene, leading to the branched products 4 and 5. Although only two examples of alkylation reactions were presented in Hiyama’s paper, it was clearly shown that, under the reaction conditions used, the insertion of Ni(0) into a C-H bond was favored over insertion into a C-F bond. This was in sharp contrast to previous studies regarding the oxidative addition of Ni(0) to polyfluorobenzenes in which the C-F bond had reacted preferentially [57,58].
Considering the high acidity of the 5-C-H bond of 2-phenyl-1,3,4-oxadiazole (6), this bond was regioselectively activated by using a catalytic amount of a Ni(cod)2 complex, together with phosphine ligands, coupled with styrenes 7 to give branched alkylated oxadiazoles 8 (Scheme 4) [59]. The bite angle of the phosphine ligand appeared to play an important role in the reaction outcome, and the Xantphos ligand was found to dramatically increase the yield of the oxadiazole products 8. For comparison, oxadiazole 6 gave with styrene only 17% of the corresponding product in the presence of PCy3 ligand, while the Xantphos ligand gave 83% of the same product under otherwise identical reaction conditions. Styrenes containing either an electron-withdrawing or an electron-donating group were comparably reactive in this coupling reaction, whereas simple aliphatic alkenes and acrylates did not yield the corresponding alkylated 1,3,4-oxadiazole products. The importance of this nickel-catalyzed alkylation with styrenes was recognized by the observed regioselectivity, which is in a stark contrast to the similar ruthenium-catalyzed reactions that yield predominantly linear coupling products [60,61]. The observed branch selectivity with styrenes in this nickel-catalyzed reaction could be explained by the preferential formation of the π-benzyl nickel intermediate A, which is not possible with simple alkenes and acrylate esters.
2.2. C-H Bond Alkylation with Alkyl Halides
In 2010, Hu et al. reported a versatile method for the alkylation of the C-H bond of N-, O- and S-containing heteroaromatics 9 with unactivated primary halides (chlorides, bromides, and iodides) (Scheme 5) [62]. This coupling reaction proceeded successfully through the cooperation of Ni(II)-complex and copper salt and showed excellent chemo- and regioselectivity in products 10 (Scheme 5). The alkylation reaction occurred exclusively at the 2-C position in both electron-poor and electron-rich heterocycles. Although the copper co-catalyst was not indispensable for the reaction, it contributed to the production of the alkylated heteroaromatics 10 in satisfactory yields. It was suggested that the copper facilitates the transmetalation of the anionic heterocyclic intermediate to the nickel center in the catalytic cycle. The Ni(II) complex C1 bearing a tridentate N,N,N-ligand served as an efficient pre-catalyst that apparently degrades into active metal-nickel particles capable of coupling heteroarenes with alkyl halides.
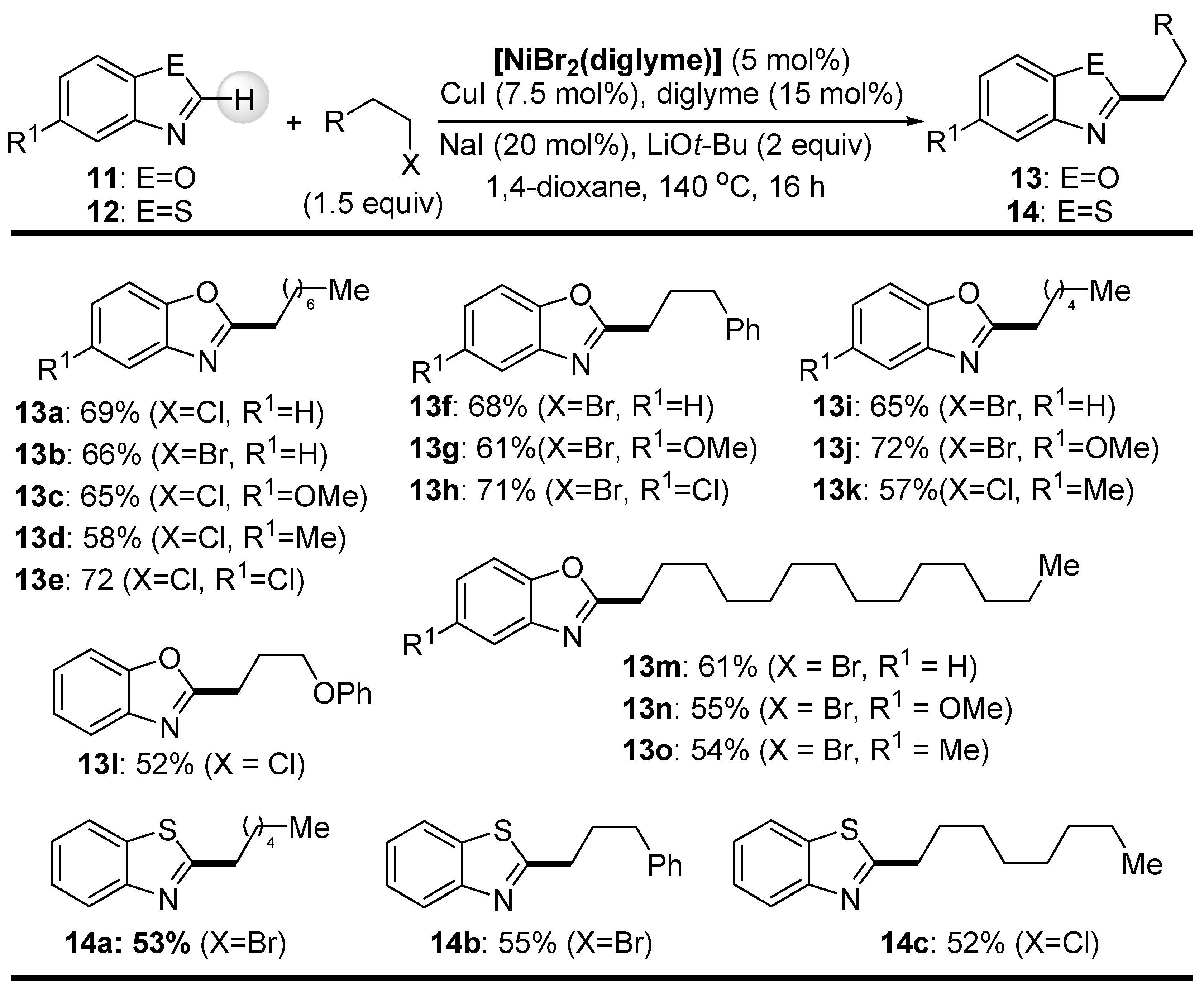
Similar to Hu’s work, Ackermann and coworkers demonstrated that benzoxazoles 11 and benzothiazoles 12 can be 2-C-alkylated with unactivated primary alkyl halides, albeit using an inexpensive NiBr2/CuI catalyst system together with a catalytic amount of diglyme for efficient C-H bond cleavage (Scheme 6) [63]. The yields of the alkylated azaheteroaromatic products 13 and 14 were generally higher than the yields obtained by the Herbert group using well-defined Ni(II) complexes of pincer ligands in the same coupling reaction. Again, both primary alkyl bromides and chlorides can be used, but NaI must be added to achieve satisfactory yields. On the other hand, the secondary alkyl bromide 2-bromohexane proved to be almost unreactive under the applied reaction conditions.
In order to gain an insight into the mode of action of the catalyst system for the alkylation reaction at hand, a few experiments were carried out (Scheme 7). The reactions of benzoxazole with cyclopropylmethyl bromide, giving 13p, and with 6-bromohex-1-ene, giving the 5-exo-cyclization product 13r, provided additional evidence for the formation of a radical intermediate involved in the catalytic cycle.
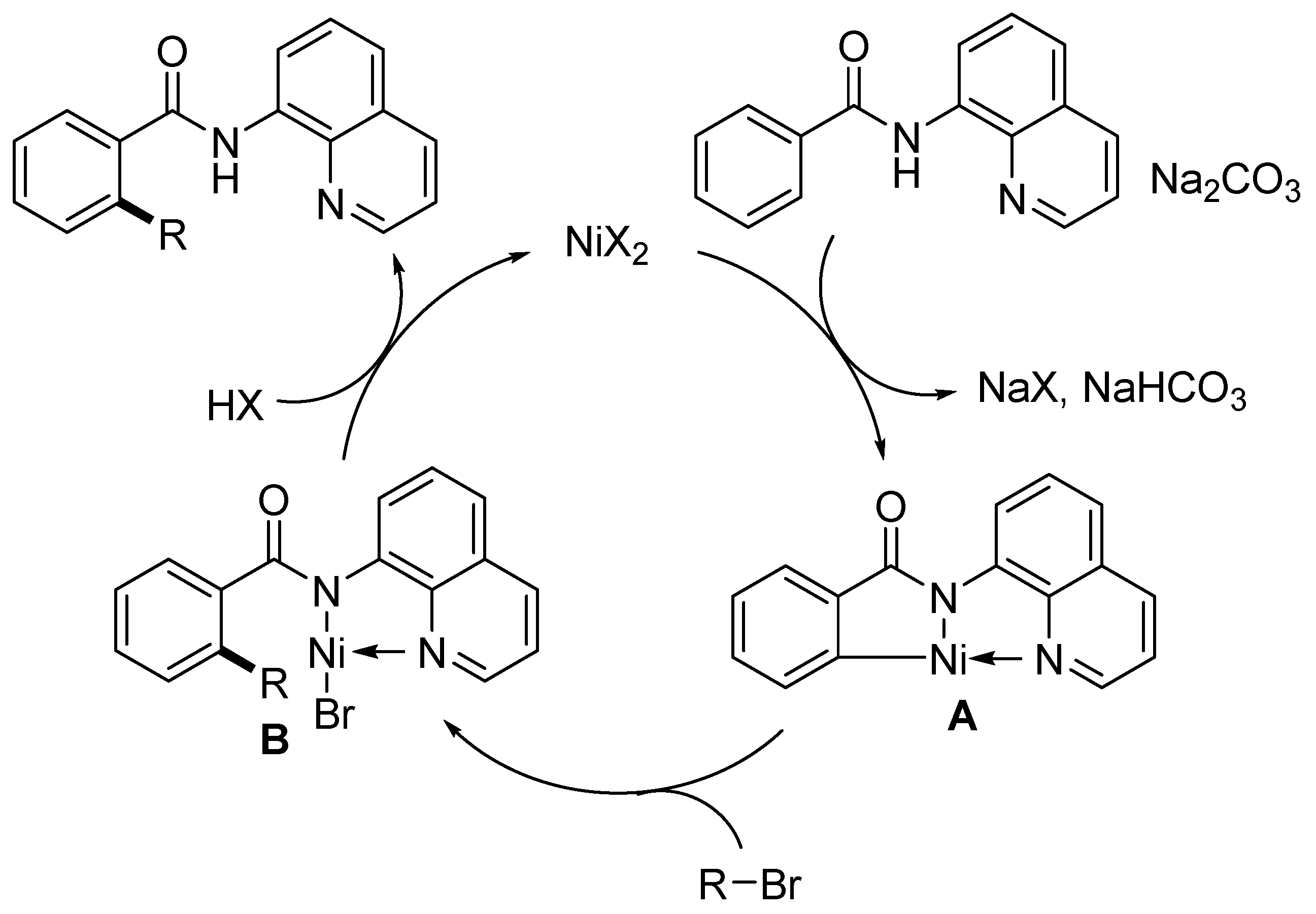
In 2013, Chatani and coworkers introduced an 8-quinolinylamine moiety as a powerful N,N-bidentate directing group in the Ru(II)-catalyzed ortho-arylation of aromatic amides [64]. This directing group was also found to be compatible with Ni(II) complexes, as the Ni(OTf)2/PPh3/Na2CO3 catalytic system efficiently performed alkylation of the ortho-C-H bond of benzamide derivatives and the β-C-H bond of α,β-unsaturated amides 15 (Scheme 8) [65]. Similarly, a thiophene ring of the heterocyclic amide was also alkylated (product 16j). The cooperation of the nickel catalyst and the N,N-bidentate-directing group appeared to be very efficient in the present direct C-H bond alkylation, as a variety of unactivated primary alkyl bromides (both simple and functionalized) were successfully used as alkylating agents. Although the use of alkyl bromides gave high yields for most amide products 16, the addition of NaI dramatically improved the isolated yields in some examples (16r and 16s). This work was a great contribution to the approach to alkyl-substituted benzene derivatives, since the metal-catalyzed alkylation of a benzene ring is otherwise limited due to the unfavorable oxidative addition of alkyl halides and, moreover, the resulting alkyl metal intermediates tend to undergo a β-hydride elimination reaction [66]. Surprisingly, α,β-unsaturated amides lacking a substituent at the α-carbon were not reactive, thus limiting the reaction to trisubstituted alkene substrates giving the corresponding products 16k–m. The authors also demonstrated that bidentate N,N-coordination by the 8-aminoquinolinyl moiety is essential for the reaction, as N-2-naphthylbenzamide and 8-quinolinyl benzoate do not give the desired alkylated products under the same reaction conditions. It has also been shown that the reaction does not proceed if no hydrogen is present on the nitrogen atom of the carboxamide group, as in substrate 15t.
Based on additional studies (H/D exchange, competition experiments), the reaction pathway starting with amide coordination to the nickel atom, followed by cyclometallation giving the intermediate A, was proposed (Scheme 9). The oxidative addition of alkyl bromide, followed by reductive elimination, yields intermediate B, which, after protonation, gives the final alkylated amide product.
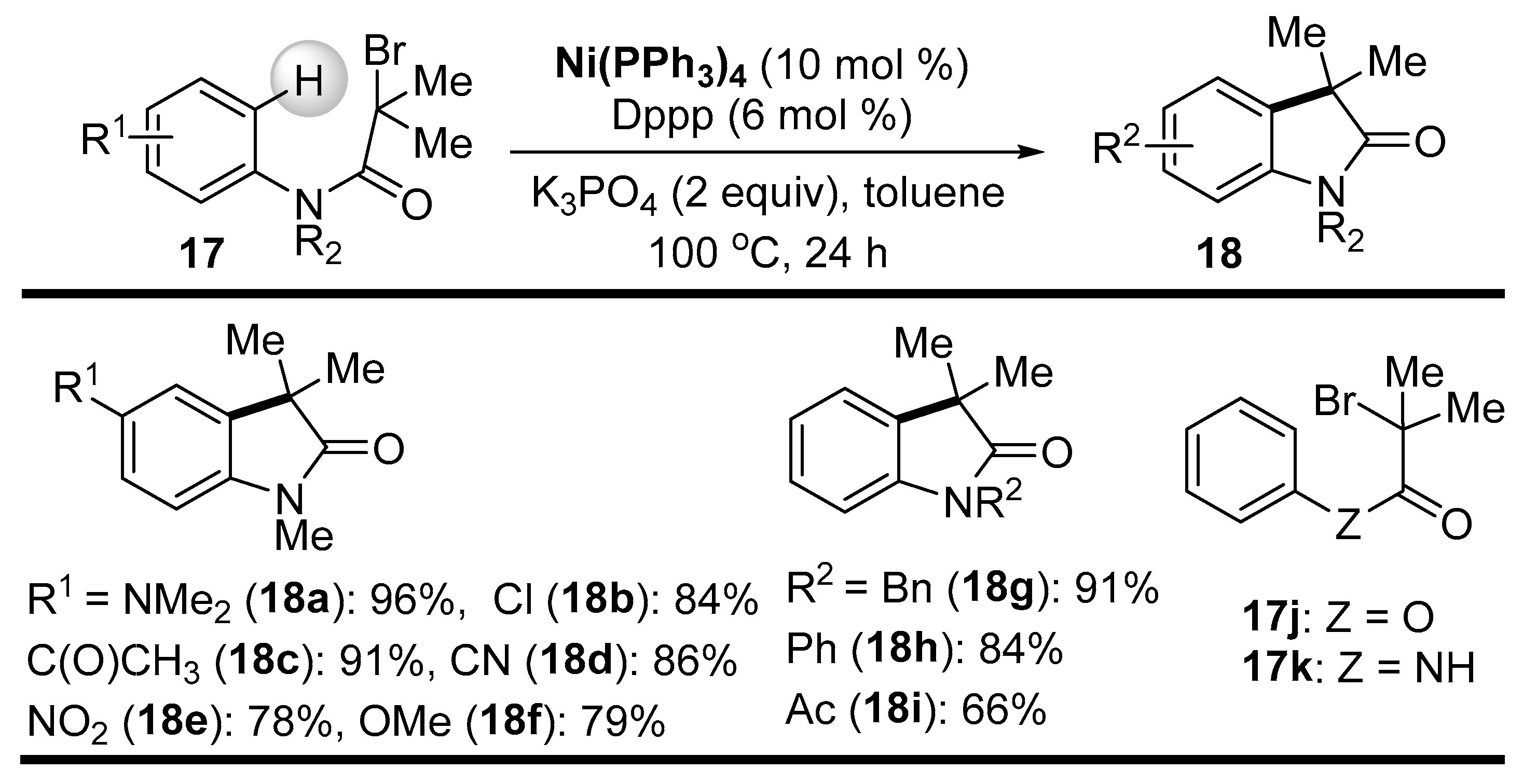
Lei and coworkers demonstrated a synthetically useful intramolecular alkylation of ortho-C-H bonds in a benzene ring of N-aryl α-bromoamide substrates 17 for the rapid preparation of indolone derivatives 18 [67]. The Ni(0) complex, together with the Dppp ligand, efficiently promoted this alkylation reaction, most likely via a radical process, as hypothesized on the basis of experiments with radical scavengers. Amide substrates 17 containing tertiary alkyl–Br bonds underwent this intramolecular cyclization smoothly and afforded the corresponding indolones 18 in about 80% yield, indicating an increased stability of the tertiary alkyl radicals (Scheme 10).
Interestingly, the analogous ester substrate 17j did not cyclize under these conditions, and, moreover, the amide 17k with an NH group was also not reactive. However, the secondary alkyl–Br bonds in the amide substrates 19 were still reactive, but the yields of products 20 decreased significantly (average yield about 50%) (Scheme 11).
It is assumed that the above mentioned cyclization reaction proceeds via a catalytic Ni(I)/Ni(II) cycle. First, a Ni(0) complex reacts with an alkyl–X bond (X = halogen) to generate a catalytically active Ni(I) species, which then reacts with an alkyl halide to give a Ni(II) and alkyl radical species A (Scheme 12). Intramolecular radical addition to the aromatic ring and subsequent oxidation by Ni(II) with simultaneous deprotonation yields the final product and regenerates the Ni(I) species.
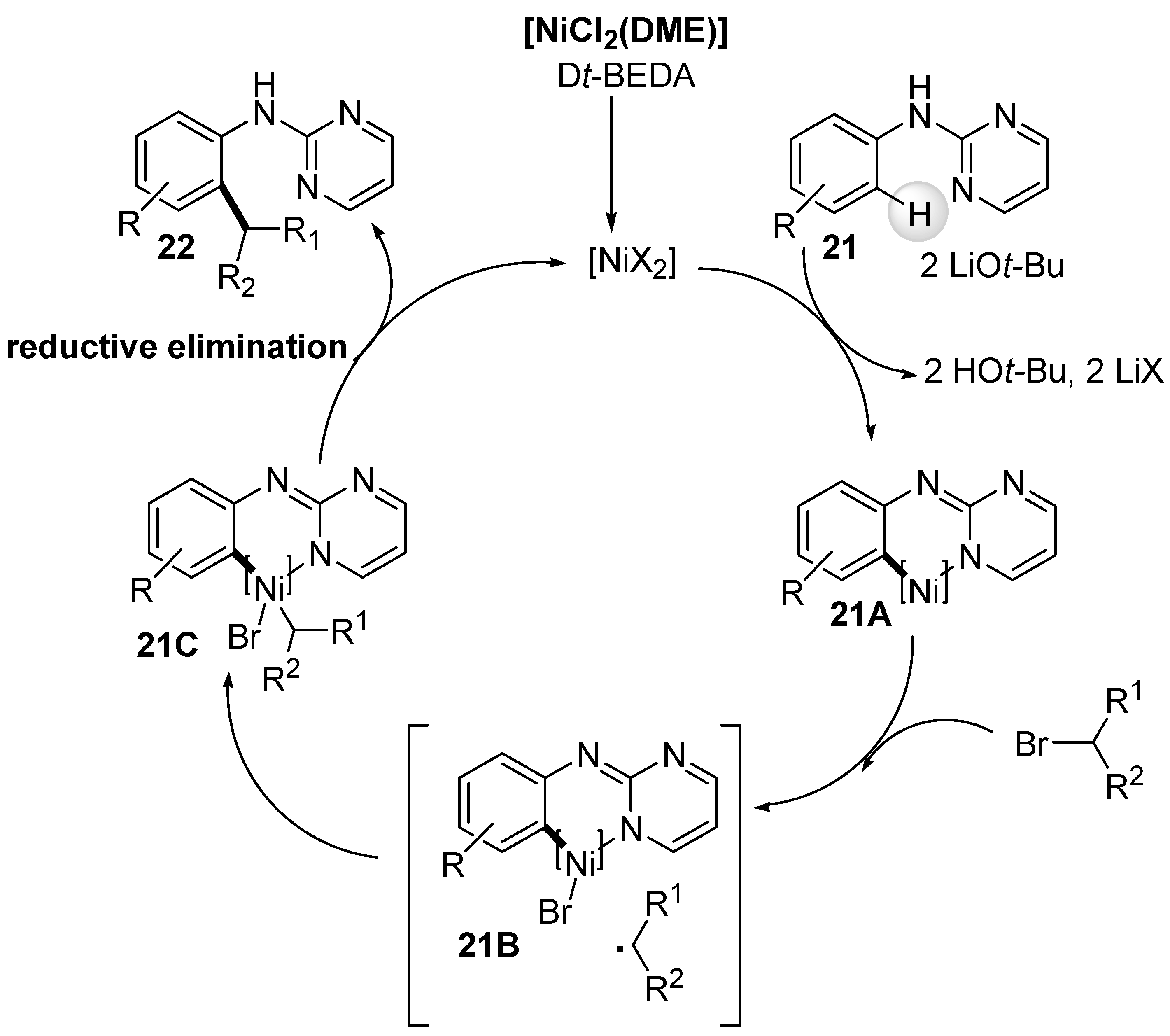
Another ortho-C-H bond alkylation, but using primary and secondary alkyl halides, was reported by Ackermann et al. [68]. They developed a protocol for the direct alkylation of anilines 21 bearing a 2-pyrimidyl moiety as a monodentate directing group on the nitrogen atom of the amino group. Interestingly, the reaction conditions introduced by Chatani et al. [66], who otherwise used the bidentate 8-quinolinylamine directing group (Scheme 8, vide supra), did not yield the desired ortho-alkylated aniline products for the nickel-catalyzed alkylation shown in Scheme 13. It was hypothesized that a vicinal diamine ligand Dt-BEDA plays an important role in the catalytic process mediated by the Ni(II) salt to achieve high yields of alkylation products 22. This could be due to the ligand-promoted formation of normally thermodynamically less stable six-membered metallacycle intermediates, such as A, in the catalytic cycle. A broad range of functionalized primary and secondary alkyl bromides can be coupled with ortho-, meta-, and para-substituted anilines with different functionalities in a highly chemo- and regioselective manner.
Based on the mechanistic studies performed, a plausible mechanism was proposed (Scheme 14) [68]. The initial [NiCl2(DME)] precatalyst reacts with Dt-BEDA ligand to form an active [NiX2] catalyst. This active catalyst initiates the catalytic cycle by reacting with 2-pyrimidinyl aniline 21 in the presence of two equivalents of LiOt-Bu as a base. Deprotonation of the aniline NH, coordination with [Ni], followed by C-H bond cleavage affords a six-membered cyclometalated complex 21A. Single electron transfer between the alkyl halide and the cyclometalated complex 21A gives intermediate 21B and an alkyl radical. The subsequent reaction affords cyclometalated complex 21C, which, upon reductive eliminations, gives rise to the alkylated 2-pyrimidinyl aniline 22 with simultaneous regeneration of the active [NiX2] catalyst (Scheme 14).
The synthetic utility of pyrimidinyl-assisted ortho-C-H alkylation of the benzene ring of anilines was demonstrated by a simple removal of the 2-pyrimidinyl directing group (Scheme 15) [68]. Heating of the alkylation products 22 in aqueous HCl under microwave conditions afforded the corresponding ortho-alkylated anilines 23 with a free amino group that can be used for further synthetic manipulations (Scheme 15).
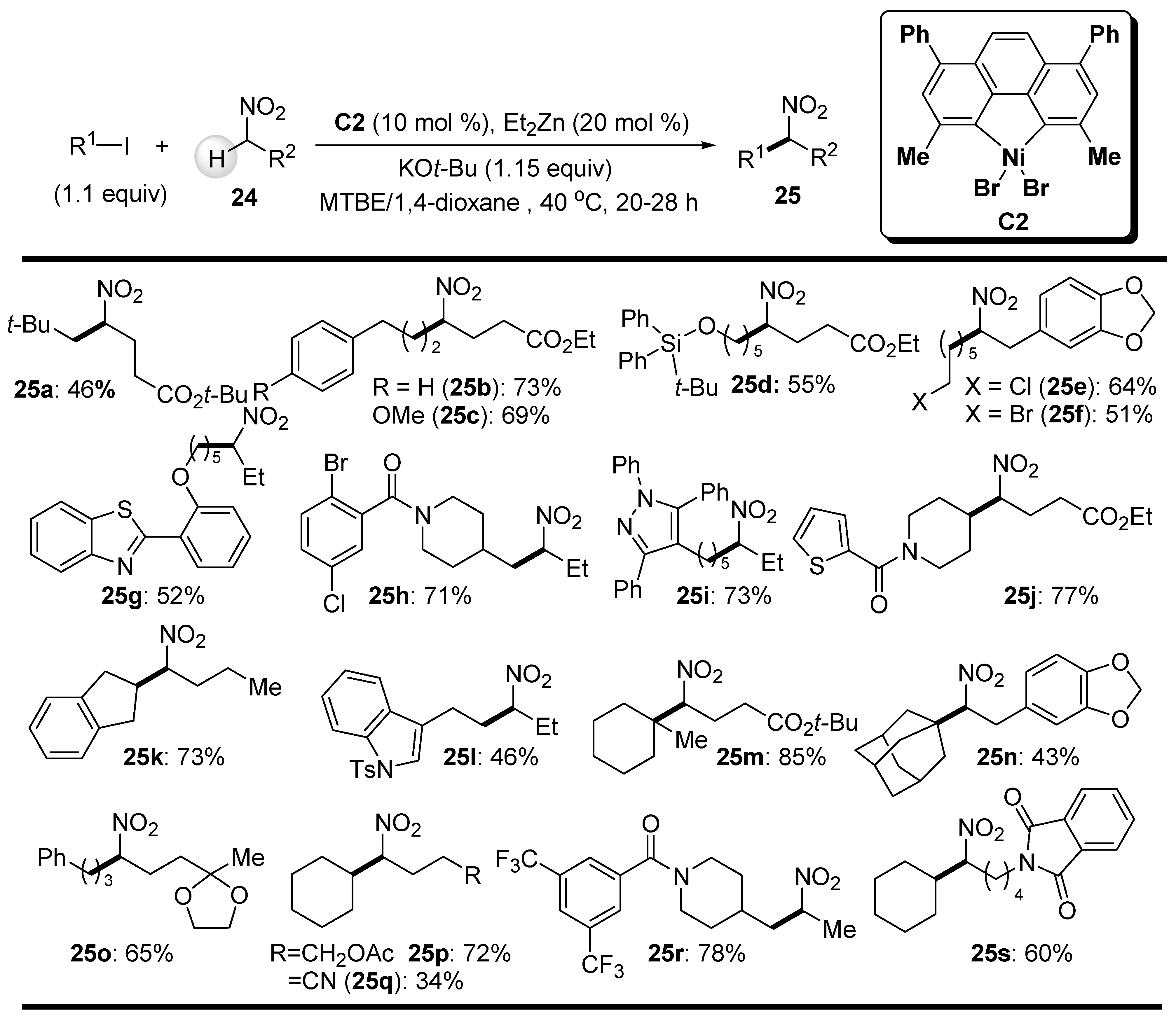
In 2017, Watson and coworkers reported the general catalytic C-H bond alkylation of nitroalkanes with unactivated alkyl halides using the single-component nickel catalyst C2 prepared from a Ni(II) precursor and bathocuproine ligand (Scheme 16) [69]. Importantly, this catalyst, together with a catalytic amount of ZnEt2 as a reducing agent, was active at a temperature of 40 °C and gave the branched nitroalkanes in moderate-to-good yields. The generality of this protocol lies in the use of primary, secondary (cyclic and acyclic) and tertiary alkyl iodides as alkylating agents with different functionalities (e.g., heteroaryl, silyl, carboxamide) in the preparation of branched nitroalkanes 25. In addition, different functional groups (e.g., ester, nitrile, imide) are well-tolerated on the nitroalkane substrates 24. Interestingly, alkyl chlorides and bromides were not affected under the applied reaction conditions, as demonstrated by the successful preparation of bromo- and chloronitroalkanes, 25e and 25f. Although a nitroalkane with a ketone group did not give the desired alkylated product, its acetal protection enabled a good yield of product 25o (Scheme 16).
The synthetic utility of this alkylation reaction was demonstrated by the preparation of a pharmaceutically relevant compound adapromine (26) (Scheme 17). First, 1-nitropropane was alkylated with 1-adamantyl iodide according to an optimized reaction protocol. The resulting product 25t was then hydrogenated under Raney nickel conditions, giving the target compound 26 in an overall yield of 52% (Scheme 17).
Punji et al. developed a synthetic method for the direct 2-C-H alkylation of N-(2-pyridyl)indoles 27 with alkyl halides in the presence of the Ni(II) catalyst C3 containing a pincer-type ligand (Scheme 18) [70]. Of the two catalysts tested, the acetate C3a was more efficient than the chloride C3b in coupling of 1-iodooctane with N-pyridylindole, giving the 2-alkylated indole 28a in a yield of 82%; the catalyst C3b gave 53% of the same product. Primary and secondary iodides and bromides were used as coupling partners, and the reaction had to be carried out at a high temperature of 150 °C. When secondary bromides were used as alkylating agents, the addition of two equivalents of KI was required to obtain satisfactory yields of the corresponding alkylated products 28l–n. Importantly, a 2-pyridyl directing group on the nitrogen atom of the indole substrates was crucial to obtain regioselectively 2-alkylated indole derivatives 28 (Scheme 18).
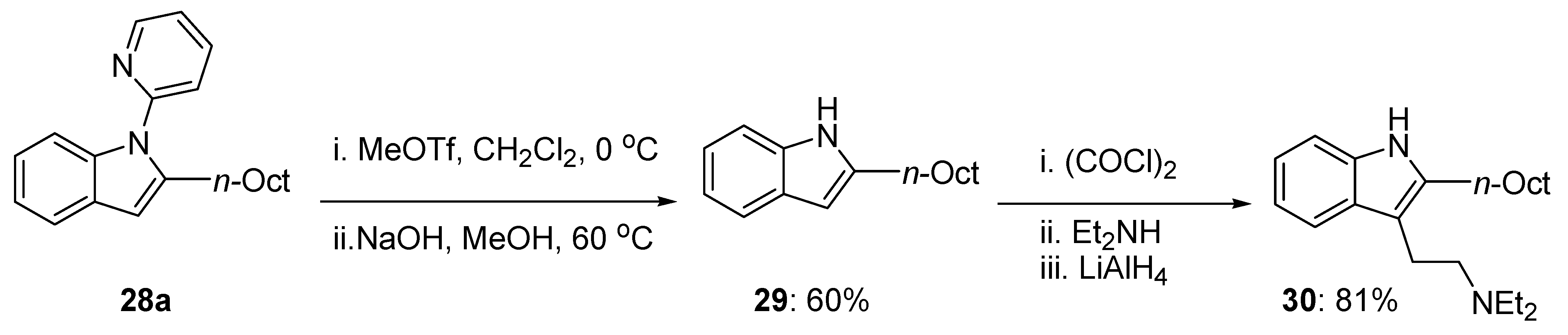
The synthetic value of the present coupling method was justified by the synthesis of the tryptamine alkaloid 30 (Scheme 19) [70]. A simple removal of the 2-pyridyl group in the starting indole 28a with MeOTf afforded the corresponding free NH-indole 29 in 60% yield. Further functionalization at the 3-C of indole 29 gave the desired tryptamine derivative in 81% yield (Scheme 19) [70].
As an improvement of their aforementioned work, Punji et al. described a milder (60 °C) yet efficient coupling of alkyl halides and 2-C-H bond not only of N-(2-pyridyl)indoles but also of N-(2-pyridyl)pyrroles 31 (Scheme 20) [71]. It was shown that the use of the nickel catalyst system NiBr2·(THF)2/Bpy, together with the strong base LiHMDS, gives the corresponding 2-alkylated indole and pyrrole derivatives 32a–k and 32l–m, respectively. Milder bases, such as KOt-Bu or K2CO3, gave only traces of the coupling product or were ineffective. Apparently, the cooperation of Ni(II) species, the ligand Bpy and a strong base is responsible for the successful coupling of indoles with alkyl halides at lower temperatures of 60 °C. Remarkably, less expensive (compared to analogous bromides or iodides) and readily available primary alkyl chlorides were used for this catalytic reaction, which gave good-to-excellent yields (up to 96%) of the 2-alkylated heteroaromatic products 32. While primary and secondary alkyl halides reacted smoothly under the applied conditions, coupling with tert-butyl chloride or 1-chloroadamantane as sterically demanding reactants was not observed. The reaction protocol tolerated a wide range of functional groups (e.g., acetal, thioether, silyl, heteroaryl, alkenyl and alkynyl) in the primary alkyl chlorides, but the electrophiles containing a -CO2R, -CN or -NO2 group decomposed under the applied reaction conditions. Surprisingly, 1,6-dichlorohexane reacted very efficiently, with only one (sp3)C-Cl under optimized conditions, giving the product 32c in high yield (80%) (Scheme 20). In addition, some mechanistic studies were carried out, which revealed that a single-electron transfer process involving the Ni(I) and Ni(III) species could occur during the catalytic cycle.
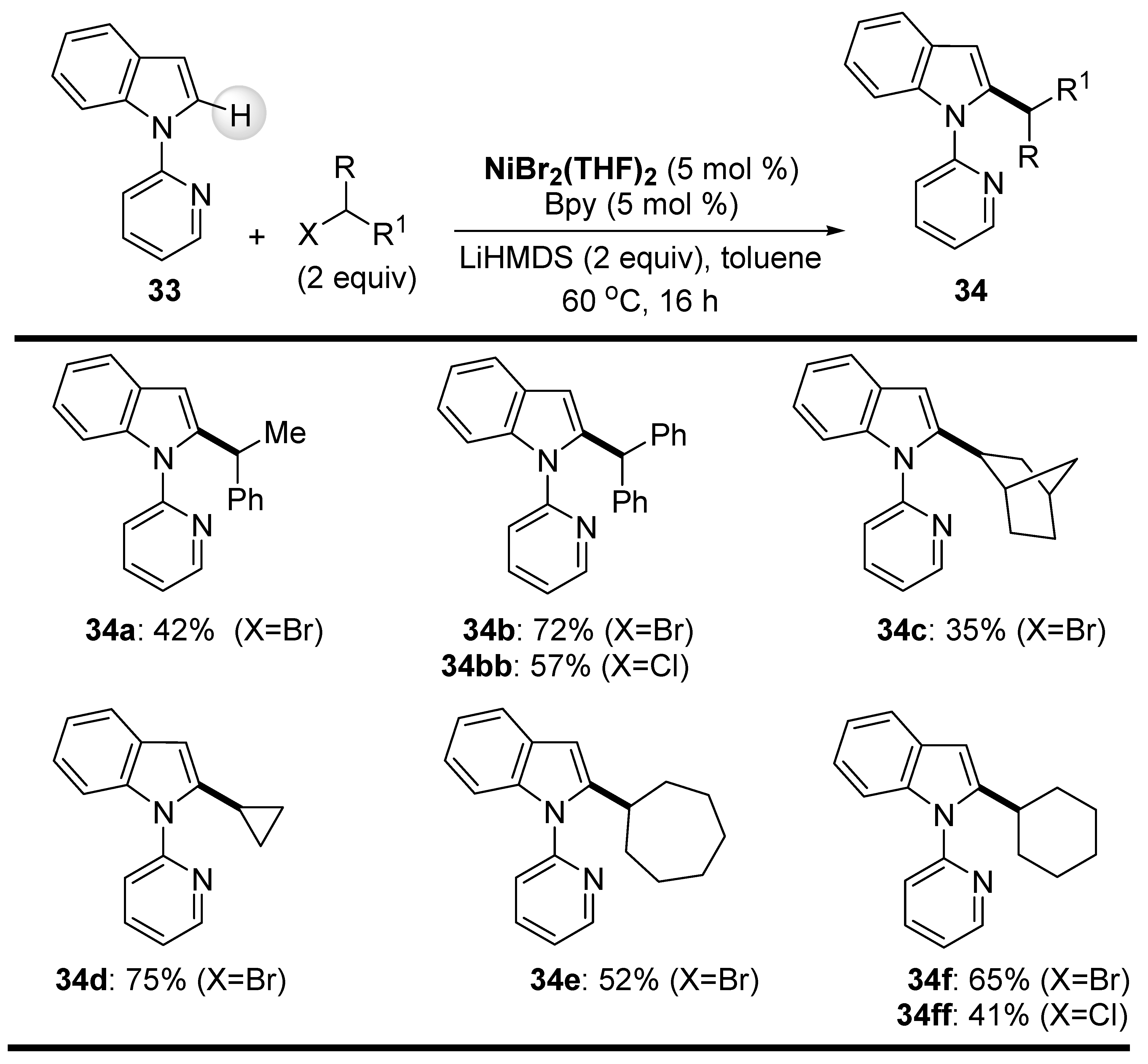
The optimized reaction conditions were also applied to the coupling reaction of secondary alkyl halides with the 2-C-H bond of N-(2-pyridyl)indole (33) (Scheme 21) [71]. Phenyl-substituted acyclic chlorides and bromides afforded products 34a and 34b in moderate-to-good yields, whereas coupling of 2-chlorobutane and 2-chloropentane with the same indole 33 afforded only trace amounts of target products. Cyclic secondary alkyl halides coupled smoothly with indole 33 and afforded 2-alkylated indoles 34d–34f in satisfactory yields. On the other hand, the reactivity of 2-bromobicyclo [2.2.1]heptane was slightly lower under the optimized conditions (35% of product 34c), probably due to the increased steric effect of this particular electrophile (Scheme 21).
After a series of mechanistic investigations, a plausible catalytic pathway was postulated (Scheme 22) [71]. The initial Ni(II) in NiBr2(THF)2 was reduced to an active Ni(I) species, [Ni(X)(bpy)], in the presence of LiHMDS, as supported by EPR and XPS studies. Additionally, 2-Pyridinyl indole 31A then reacted with the resulting low-valent Ni(I) in the presence of LiHMDS to produce cyclometalated Ni-complex 31B. As a rate-determining step, the alkyl chloride underwent a radical formation triggered by the Ni-complex 31B, giving rise to the Ni species 31C and the alkyl radical. The decoordination between the Lewis basic N atom in the pyridinyl moiety and the Lewis acidic Ni, followed by the formation of the Ni–C bond between Ni and the radical-bearing C in the alkyl radicals, gave the Ni intermediate 31D. Final reductive elimination furnished the alkylated 2-pyridinyl indole 32 with concomitant regeneration of the active low-valent Ni(I) catalyst (Scheme 22) [71].
The authors also investigated this coupling strategy in the preparation of symmetrical and, in particular, unsymmetrical bis(indolyl)alkane scaffolds using a one-pot synthetic strategy (Scheme 23). Treatment of the starting indole 33 (2 equiv.) with 1-bromo-4-chlorobutane (0.5 equiv.) gave a symmetrical bis(indolyl)butane 35, while an unsymmetrical analog 37 was prepared by a sequential reaction of 1-bromo-4-chlorobutane (1.3 equiv.) with indoles 33 (1 equiv.) and 36 (2 equiv.); additionally, the monoindolation product was not isolated (Scheme 23).
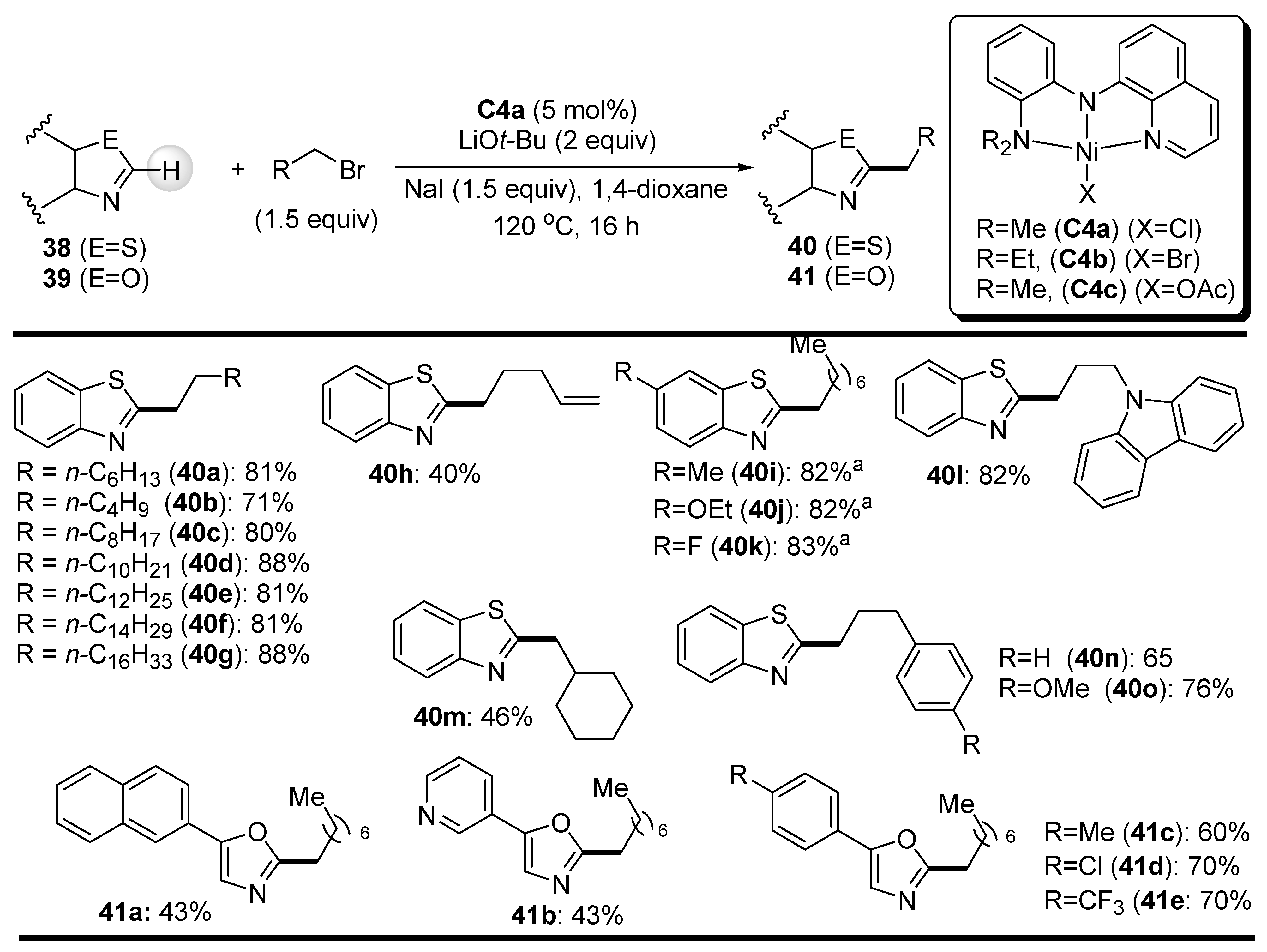
Punji et al. extended the scope of direct 2-C-alkylation of fused azaheteroarenes (see Scheme 24) by using well-defined Ni(II) complexes of tridentate N,N,N-pincer ligands. Thus, the complexes C4 of quinoline-based ligand were investigated in the catalytic C-H bond alkylation of benzothiazoles 38 and oxazoles 39 with primary alkyl halides (Scheme 22) [72]. Complex C4a showed the best performance in the model reaction of 1-iodooctane with benzothiazole and proved to be a very robust catalyst in the present coupling reaction, as it can be reused five times without a significant decrease in its activity. For example, the yield of product 40a in the reaction of benzothiazole with 1-iodooctane decreased from 92% to 85% under optimized conditions when moving from the first to the fifth cycle. In particular, the extent of the alkylation reaction with readily available primary alkyl bromides was investigated, but in order to obtain satisfactory yields of products 40 and 41, NaI (1.5 equiv.) was added to the reaction mixture (Scheme 24).
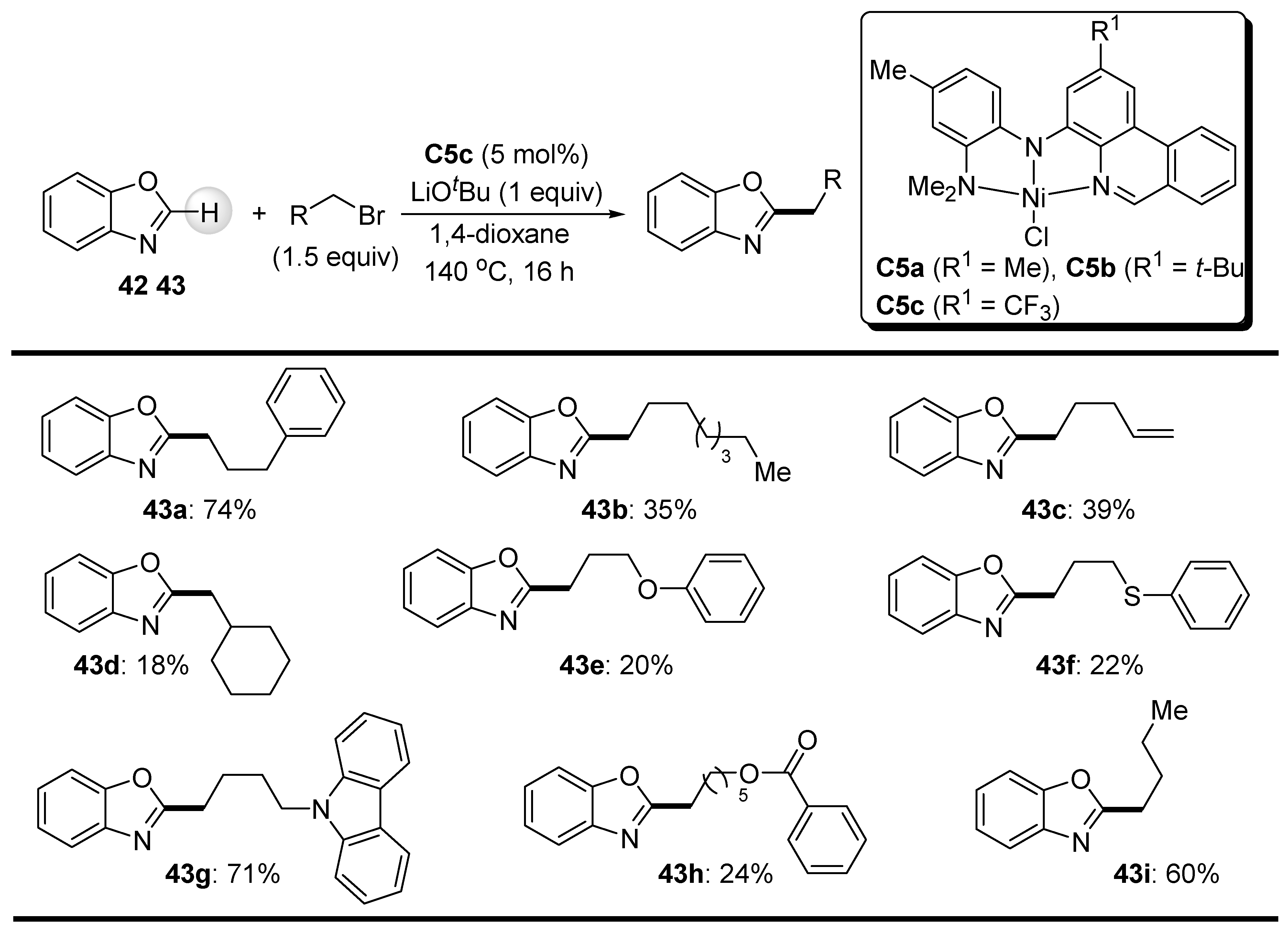
As an exploration of the direct alkylation method mentioned above (Punji’s work), nickel complexes C5 ligated with NNN-tridentate but phenanthridine-based ligands proved to be satisfactorily efficient catalysts in the functionalization of benzoxazole (42) with unactivated primary alkyl bromides in a highly site-selective manner, as reported by Herbert et al. (Scheme 25) [73]. Of the nickel complexes tested, complex C5c showed the best catalytic performance in this coupling reaction. Although the yields of 2-alkylated benzoxazole derivatives 43 were generally not high, the examples shown in Scheme 25 represent a significant contribution to nickel-promoted functionalization of C-H bonds, particularly because well-defined Ni(II) complexes were used and pincer-like ligands can efficiently stabilize a catalytically active nickel center.
Recently, Martin et al. discovered that unactivated α-olefins 44 and unactivated alkyl bromides elegantly couple under light-induced nickel catalysis to readily form a C(sp3)–C(sp3) linkage between the α-carbon atom of olefins and an alkyl moiety of alkyl halides by simultaneous hydrogen atom transfer at the allylic C(sp3)–H bond [74]. This coupling protocol proved to be widely applicable, as primary and secondary alkyl bromides with different functionalities (e.g., acetal, ester, alcohol, carbamate, sulfonamide, alkenyl, alkynyl) can be used to chemoselectively obtain moderate-to-good yields of internal olefin products 45 (Scheme 26). The catalyst system NiBr2·glyme/L1/Li2CO3, used together with the photoactive iridium complex C6, allowed a smooth alkylation reaction with primary alkyl bromides at 34 °C, obtaining products 45 in good yields (Scheme 24). Of particular interest is the use of pyrazole-containing primary alkyl bromide, which does not interfere with the formation of the alkylated product 45f, indicating that the pyrazole ring does not compete with the ligand L1 for binding to the nickel center in the catalytic cycle. It is worth noting that this protocol was also suitable for alkyl bromides containing amino acid, saccharide and steroid fragments (with free alcohol groups), resulting in the products 45m–o (Scheme 26).
On the other hand, a protocol based on the NiBr2·glyme/L2/K2CO3 catalytic system, together with a metal-free photoredox catalyst C7, was more suitable for alkylation with secondary alkyl bromides (acyclic and cyclic) in regard to giving olefinic products 46 (Scheme 27). Remarkably, aldehyde 48 and ketone 49 were obtained in good yields when the parent alkyl bromides were coupled with allylic alcohol 47 as an olefinic coupling partner under the same reaction conditions (Scheme 27). Although mixtures of E/Z-isomeric olefin products were isolated in the present coupling reactions, the developed protocol allowed the synthesis of densely functionalized compounds from simple starting materials.
2.3. Miscellaneous C-H Bond Alkylations
Martin et al. presented an interesting deaminative alkylation of unactivated olefins with pyridinium salts 50 (which can be easily prepared from the corresponding amines and pyrylium salts) via their (sp3)C–N bond cleavage (Scheme 28) [75]. This unique coupling reaction proceeds as a 1,2-hydroalkylation of the C=C double bond of the olefins to generate a new (sp3)C–(sp3)C bond in the products 51 or 52 in the presence of catalytic amounts of Ni(II) salt/ligand, a hydride source ((EtO)2MeSi–H or (EtO)3Si–H) and Na2HPO4 as base (Scheme 28). It was found that, for the alkylation of terminal olefins, the NiBr2·glyme complex, together with the bipyridine ligand L3, is the privileged catalyst system that operates successfully at 40 °C with excellent anti-Markovnikov selectivity (Scheme 26). It is worth mentioning that only the mixture of NMP and 1,4-dioxane as a solvent system gave the best reaction outcome, as a single solvent gave a lower yield (Scheme 28).
On the other hand, the stabilization of the catalytically active nickel center (originating from NiI2) in the deaminative alkylation of internal olefins was achieved by the oxazoline-pyridine ligand L4 (Scheme 29). Although internal olefins were used as substrates, (sp3)–C alkylation occurred at a distant primary (sp3)C-H site, which gave products 52 regardless of the position of the double bond. In the alkylation of internal olefins, a DMSO/1,4-dioxane solvent system was used, while the reaction took place at a lower temperature of 35 °C (Scheme 29).
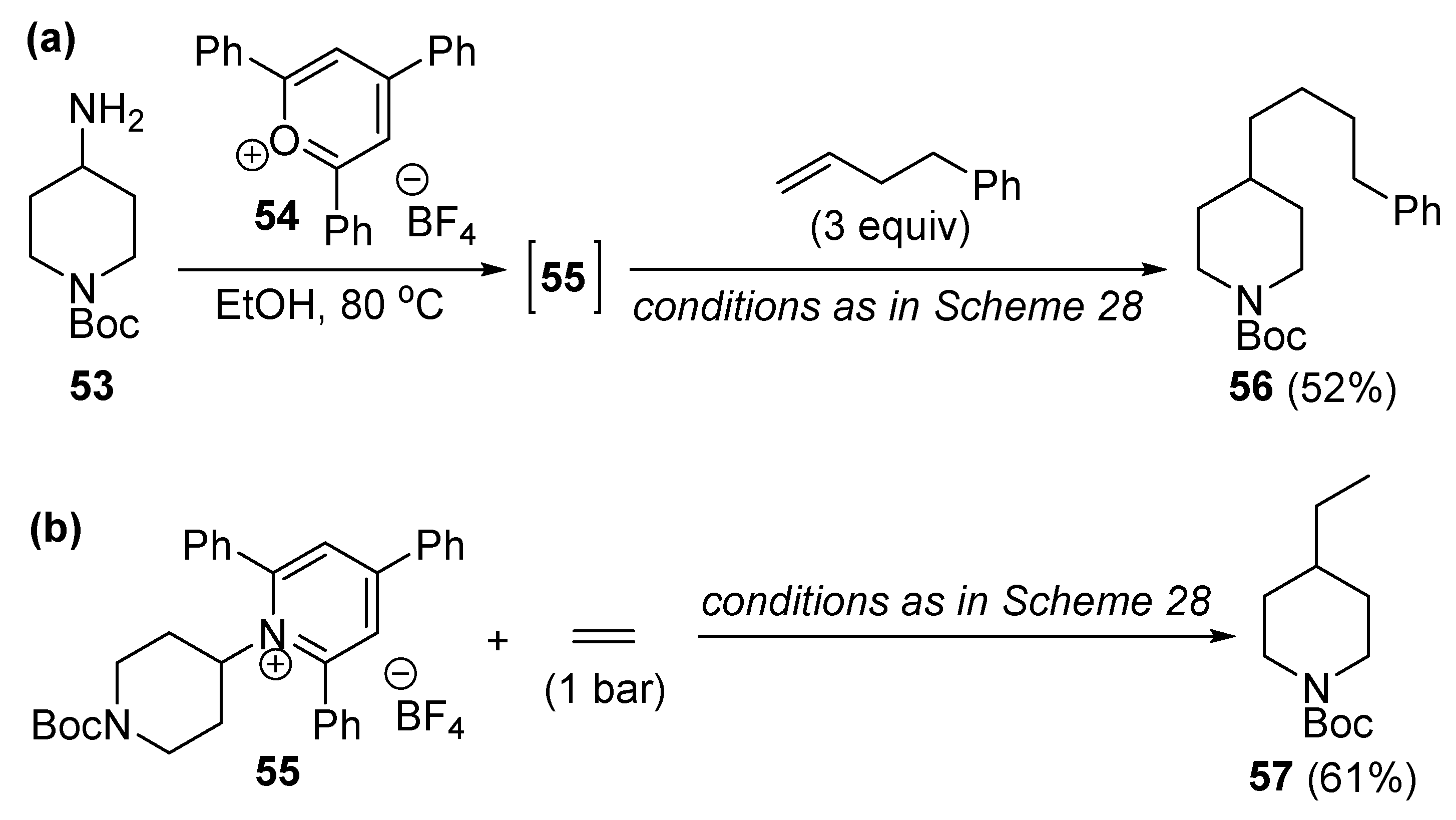
As far as synthetic applications are concerned, the authors have shown (albeit in only one case) that the deaminative alkylation of olefins can also be achieved with the in situ formed pyridinium salt 55 from the parent piperidinyl amine 53 and pyrylium salt 54 using a one-pot synthetic strategy (Scheme 30a). It is worth noting that ethylene, as an industrially important chemical, can also be successfully used as a coupling partner under atmospheric pressure to produce ethylated molecular scaffolds (product 57, Scheme 30b).
Finally, it was shown in several cases that the developed protocol can be applied for the late-stage selective functionalization of substrates derived from natural products, e.g., camphene, (−)-β-pinene, linalol, D-galactose, estrone, and (+)-valencene (products 58–63, Scheme 31).
2.4. Asymmetric C-H Bond Alkylation
Recently, Shi’s group published the then first enantioselective C-H bond alkylation of pyridines 64 with β-substituted styrenes 65, which was realized using a bimetallic nickel-aluminum catalyst system with an N-heterocyclic ligand L5 (Scheme 32) [76]. The reaction proceeded formally as an asymmetric addition of a para-C-H bond of pyridine 64 across the C=C double bond of the alkene, leading to 1-aryl-1-pyridylalkanes 66. Enantiocontrol was provided by a C2-symmetric chiral acenaphthene-type NHC ligand L5 bearing bulky 3,5-Xylyl groups. It is noteworthy that the present activation of the pyridyl C-H bond was not possible without the Lewis acid methylaluminium bis(2,6-di-tert-butyl-4-methylphenoxide) (MAD) or with smaller Lewis acids, such as AlMe3, AlEt3 or Al(iBu)3. It is suggested that the complete para-regioselectivity in pyridine substrates is due to the steric effect of MAD in a Lewis acid–base-MAD–pyridine adduct, which prevents the functionalization of the pyridyl ortho-C-H bond. As shown in Scheme 30, a wide range of substituted styrene and pyridine coupling partners were successfully utilized in this catalytic reaction, leading to 1,1-diarylalkanes with pyridyl moieties in high yields and good-to-excellent enantioselectivities at a temperature of 50 °C. Interestingly, a ferrocene-containing alkene also reacted smoothly under slightly modified conditions (3 equiv. of alkene, 60 °C, 48 h), affording the alkylated pyridine derivative 66l with an exceptionally high enantioselectivity (97% ee). In addition, quinoline substrates also afforded the desired 4-C-H alkylated products 67 and 68, albeit in moderate yield, with good enantioselectivity (~65% ee) (Scheme 32).
Mechanistic investigations were carried out for the Ni-catalyzed C-H alkylation of pyridines with styrenes [76]. Based on previous findings and calculated Gibbs energy profiles, the reaction mechanism is believed to begin with substrate coordination, proceeds through ligand to ligand hydrogen transfer (LLHT), before ending with reductive elimination. A plausible mechanism was postulated on the basis of energy calculations (Scheme 31). The mechanism given below applies to the energetically more favorable S isomers of the intermediates and transition states in the energy profile [76]. The Ni precatalyst, Ni(cod)2, undergoes NHC ligand L5 exchange with cod to give an active Ni catalyst, [Ni(cod)(NHC)]. The active Ni catalyst then coordinates with the trans styrene 65a to form intermediate IA. The intermediate IA then coordinates with the Lewis adduct formed by the reaction between the Lewis basic pyridine 64 and the Lewis acidic MAD (Scheme 32, vide supra) to give intermediate IB. The LLHT then takes place through transition state TS1, which gives rise to the intermediate IC. The intermediate IC undergoes reductive elimination through transition state TS2 to form the intermediate ID, which then releases the pyridine alkylated product 66a by ligand exchange along with pyridine-MAD Lewis adduct, simultaneously regenerating the IA (Scheme 33).
3. Conclusions
Ni-catalyzed alkylation of C-H bonds is an attractive strategy for the direct functionalization of inert C-H bonds of various substrates. One advantage is the use of Ni as a first-row transition metal, which occurs in larger quantities in the earth’s crust and is more environmentally friendly than the second-row transition metals. Another advantage is the possibility of incorporating alkyl groups into valuable molecules, which allows access to natural product frameworks with differently functionalized alkyl groups. This review focuses on the Ni-catalyzed C-H bond alkylation reactions of various carbocyclic and heterocyclic substrates with alkyl halides and olefins. The alkylation reactions covered include, for example, intramolecular alkylation for the rapid construction of indolone scaffolds. The Ni-catalyzed C-H bond alkylation of aromatic and unsaturated amides with alkyl halides and of nitroalkanes with unactivated olefins is also covered. The regioselectivity of C-H bond alkylation in unactivated substrates was, in many cases, ensured by chelation assistance with monodentate pyridyl and pyrimidyl or bidentate quinolinylamine directing groups. The unique enantioselective alkylation of pyridines with alkenes paves the path for future Ni-catalyzed asymmetric alkylations.
Funding
This review received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The financial support from the Slovenian Research Agency through grant P1-0179 is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| Ac | Acyl |
| Ar | Aryl |
| Bn | Benzyl |
| Boc | tert-Butyloxycarbonyl |
| Bpy | 2,2-Bipyridine |
| Bu | Butyl |
| Cbz | Benzyloxycarbonyl |
| Cod | 1,5-Cyclooctadiene |
| Cp | Cyclopentadienyl |
| Cy | Cyclohexyl |
| Cyp | Cyclopentyl |
| DCM | Dichloromethane |
| diglyme | bis(2-methoxyethyl) ether |
| DMA | Dimethylacetamide |
| DME | Dimethoxyethane |
| DMSO | Dimethylsulfoxide |
| dppp | 1,3-Bis(diphenylphosphino)propane |
| DtBEDA | N1,N2-di-tert-butylethane-1,2-diamine |
| ee | enantiomeric excess |
| equiv | Equivalent |
| Et | Ethyl |
| glyme | Dimethoxyethane |
| Hept | Heptyl |
| Hex | Hexyl |
| HMDS | Hexamethyldisilazide |
| iPr | iso-Propyl |
| L | Ligand |
| LED | Light-emitting diode |
| Me | Methyl |
| MTBE | Methyl tert-butyl ether |
| NHC | N-Heterocyclic carbine |
| NMP | N-Methyl-2-pyrrolidone |
| Ph | Phenyl |
| Pr | Propyl |
| RT | Room Temperature |
| TBME | tert-Butyl methyl ether |
| TBS | tert-Butyldimethylsilyl |
| Tf | Triflyl |
| THF | Tetrahydrofuran |
| TM | Transition meta |
| TMS | Trimethylsilyl |
| Xantphos | (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) |
| Xylyl | Dimethylphenyl |
| μW | Microwave |
References
- Shilov, A.E.; Shul’pin, G.B. Activation of C-H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, F.; Chatani, N. Catalytic Methods for C-H Bond Functionalization: Application in Organic Synthesis. Adv. Synth. Catal. 2003, 345, 1077–1101. [Google Scholar] [CrossRef]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C-H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Das, A.; Chatani, N. Strategic Evolution in Transition Metal-Catalyzed Directed C-H Bond Activation and Future Directions. Coord. Chem. Rev. 2021, 431, 213683–213719. [Google Scholar] [CrossRef]
- Al Mamari, H.H.; Štefane, B.; Žugelj, H.B. Metal-Catalyzed C-H Bond Functionalization of Phenol Derivatives. Tetrahedron 2020, 76, 130925–130936. [Google Scholar] [CrossRef]
- Docherty, J.H.; Lister, T.M.; Mcarthur, G.; Findlay, M.T.; Domingo-Legarda, P.; Jacob Kenyon, J.; Choudhary, S.; Igor Larrosa, I. Transition-Metal-Catalyzed C-H Bond Activation for the Formation of C-C Bonds in Complex Molecules. Chem. Rev. 2023, 123, 7692–7760. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.; Faber, T.; Glorius, F. C-H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 2, 245–261. [Google Scholar] [CrossRef]
- Omae, I. Intramolecular five-membered ring compounds and their applications. Coord. Chem. Rev. 2004, 248, 995–1023. [Google Scholar] [CrossRef]
- Rouquet, G.N.; Chatani, N. Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnio, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A Comprehensive Overview of Directing Groups Applied in Metal-Catalyzed C-H Functionalization Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef]
- de Meijere, A.; Bräse, S.; Oestreich, M. (Eds.) Metal Catalyzed Cross-Coupling Reactions and More; Wiley: Hoboken, NJ, USA, 2013; Volume 3. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Del Zotto, A.; Marco Zecca, M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef]
- Campeau, L.-C.; Hazari, N. Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 2019, 38, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium-Catalyzed Transformations of Alkyl C-H Bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. Controlling Site Selectivity in Palladium-Catalyzed C-H Bond Functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef]
- Bay, K.L.; Yang, Y.-F.; Houk, K.N. Multiple Roles of Silver Salts in Palladium-Catalyzed C-H Activations. J. Organomet. Chem. 2018, 864, 19–25. [Google Scholar] [CrossRef]
- Colby, D.A.; Tsai, A.S.; Bergman, R.G.; Ellman, J.A. Rhodium Catalyzed Chelation-Assisted C-H Bond Functionalization Reactions. Acc. Chem. Res. 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Chatani, N. Rhodium-Catalyzed C(sp2)- or C(sp3)–H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 8304–8329, Erratum in Angew. Chem. 2019, 131, 8390–8416. [Google Scholar] [CrossRef]
- Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Direct Functionalization of Nitrogen Heterocycles via Rh-Catalyzed C-H Bond Activation. Acc. Chem. Res. 2008, 41, 1013–1025. [Google Scholar] [CrossRef]
- Vasquez-Cespedes, S.; Wang, X.; Glorius, F. Plausible Rh(V) Intermediates in Catalytic C-H Activation Reactions. ACS Catal. 2018, 8, 242–257. [Google Scholar] [CrossRef]
- Song, G.; Li, X. Substrate Activation Strategies in Rhodium(III)-Catalyzed Selective Functionalization of Arenes. Acc. Chem. Res. 2015, 48, 1007–1020. [Google Scholar] [CrossRef]
- Singh, K.S. Recent Advances in C-H Bond Functionalization with Ruthenium-Based Catalysts. Catalysts 2019, 9, 173. [Google Scholar] [CrossRef]
- Dana, S.; Yadav, M.R.; Sahon, A.K. Ruthenium-Catalyzed C-N and C–O Bond-Forming Processes from C-H Bond Functionalization. Top. Organomet. Chem. 2016, 55, 189–215. [Google Scholar] [CrossRef]
- Ruiz, S.; Villuendas, P.; Urriolabeitia, E.P. Ru-catalyzed C-H functionalizations as a tool for selective organic synthesis. Tetrahedron Lett. 2016, 57, 3413–3432. [Google Scholar] [CrossRef]
- Li, B.; Dixneuf, P.H. Ruthenium(II)-Catalyzed sp2 C-H Bond Functionalization by C-C Bond Formation. Top. Organomet. Chem. 2015, 48, 119–193. [Google Scholar] [CrossRef]
- Ackermann, L.; Vicente, R. Ruthenium-Catalyzed Direct Arylations through C-H Bond Cleavages. Top. Curr. Chem. 2010, 292, 211–229. [Google Scholar] [CrossRef]
- Dixneuf, P.H.; Cadierno, V. (Eds.) Metal-Catalyzed C-H Bond Activation and C-C Bond Formation in Water; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Li, B.; Dixneuf, P.H. sp2 C-H bond activation in water and catalytic crosscoupling reactions. Chem. Soc. Rev. 2013, 42, 5744–5767. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Chelation-Assisted Nickel-Catalyzed C-H Functionalizations. Trends Chem. 2019, 1, 524–539. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Nickel-Catalyzed C-H Functionalization Using A Non-directed Strategy. Chem 2020, 6, 1056–1081. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Xia, Y.-N.; Shi, B.-F. Ni-Catalyzed Chelation-Assisted Direct Functionalization of Inert C-H Bonds. Chin. J. Chem. 2020, 38, 635–662. [Google Scholar] [CrossRef]
- Cano, R.; Mackey, K.; McGlacken, G.P. Recent Advances in Manganese-Catalyzed C-H Activation: Scope and Mechanism. Catal. Sci. Technol. 2018, 8, 1251–1266. [Google Scholar] [CrossRef]
- Liu, W.; Ackermann, L. Manganese-Catalyzed C-H Activation. ACS Catal. 2016, 6, 3743–3752. [Google Scholar] [CrossRef]
- Lanzi, M.; Cera, G. Iron-Catalyzed C-H Functionalizations under Triazole-Assistance. Molecules 2020, 25, 1806. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C-H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Prakash, S.; Kuppusamy, R.; Cheng, C.H. Cobalt-Catalyzed Annulation Reactions via C-H Bond Activation. ChemCatChem 2018, 10, 683–705. [Google Scholar] [CrossRef]
- Wang, S.; Chen, S.-Y.; Yu, X.-Q. C-H Functionalization by High-Valent Cp*Co(iii) Catalysis. Chem. Commun. 2017, 53, 3165–3180. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Matsunaga, S. High-Valent Cobalt-Catalyzed C-H Bond Functionalization. Adv. Organomet. Chem. 2019, 68, 197–247. [Google Scholar] [CrossRef]
- Baccalini, A.; Vergura, S.; Dolui, P.; Zanoni, G.; Maiti, D. Recent Advances in Cobalt-Catalyzed C-H Functionalizations. Org. Biomol. Chem. 2019, 17, 10119–10141. [Google Scholar] [CrossRef]
- Moselage, M.; Lie, J.; Ackermann, L. Cobalt-Catalyzed C-H Activation. ACS Catal. 2016, 6, 498–525. [Google Scholar] [CrossRef]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Erratum: Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef]
- Ge, S.; Hartwig, J.F. Highly Reactive, Single-Component Nickel Catalyst Precursor for Suzuki–Miyuara Cross-Coupling of Heteroaryl Boronic Acids with Heteroaryl Halides. Angew. Chem. Int. Ed. 2012, 51, 12837–12841. [Google Scholar] [CrossRef]
- Ramgren, S.D.; Hie, L.; Ye, Y.; Garg, N.K. Nickel-Catalyzed Suzuki–Miyaura Couplings in Green Solvents. Org. Lett. 2013, 15, 3950–3953. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Kleiman, J.P.; Dubeck, M. The Preparation of Cyclopentadienyl [o-(Phenylazo)Phenyl]Nickel. J. Am. Chem. Soc. 1963, 85, 1544–1545. [Google Scholar] [CrossRef]
- Jagtap, R.A.; Punji, B. Nickel-Catalyzed C H Bond Functionalization of Azoles and Indoles. Chem. Rec. 2021, 21, 3573. [Google Scholar] [CrossRef] [PubMed]
- Harry, N.A.; Saranya, S.; Ujwaldev, S.M.; Anilkumar, G. Recent advances and prospects in nickel-catalyzed C-H activation. Catal. Sci. Technol. 2019, 9, 1726–1743. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Muto, K.; Itami, K. Recent Progress in Nickel-Catalyzed Biaryl Coupling. Eur. J. Org. Chem. 2013, 2013, 19–30. [Google Scholar] [CrossRef]
- Hu, F.; Shen, Y.-B.; Wang, L.; Li, S.-S. Merging dearomatization with redox-neutral C(sp3)–H functionalization via hydride transfer/cyclization: Recent advances and perspectives. Org. Chem. Front. 2022, 9, 5041–5052. [Google Scholar] [CrossRef]
- Dong, Y.; Hu, F.; Wu, H.; Guo, F.-W.; Wang, L.; Du, F.-Y.; Li, S.-S. Controllable Synthesis of N-Heterocycles via Hydride Transfer Strategy-Enabled Formal [5 + 1] and [5 + 2] Cyclizations. Org. Lett. 2024, 26, 332–337. [Google Scholar] [CrossRef]
- Guo, W.; Wang, Q.; Zhu, J. Visible light photoredox-catalysed remote C-H functionalisation enabled by 1,5-hydrogen atom transfer (1,5-HAT). Chem. Soc. Rev. 2021, 50, 7359–7377. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, Z.; Hu, Q.; Elsaid, M.; Liu, C.; Chen, J.; Ge, H. Recent Strategies in Nickel-Catalyzed C-H Bond Functionalization for Nitrogen-Containing Heterocycles. Catalysts 2022, 12, 1163. [Google Scholar] [CrossRef]
- Clement, N.D.; Cavell, K.J. Transition-Metal-Catalyzed Reactions Involving Imidazolium Salt/N-Heterocyclic Carbene Couples as Substrates. Angew. Chem. Int. Ed. 2004, 43, 3845–3847. [Google Scholar] [CrossRef] [PubMed]
- Nakao, Y.; Kashihara, N.; Kanyiva, K.S.; Hiyama, T. Nickel-catalyzed alkenylation and alkylation of fluoroarenes via activation of C-H bond over C-F bond. J. Am. Chem. Soc. 2008, 130, 16170–16171. [Google Scholar] [CrossRef] [PubMed]
- Keen, A.L.; Johnson, S.A. Nickel(0)-Catalyzed Isomerization of an Aryne Complex: Formation of a Dinuclear Ni(I) Complex via C-H Rather than C-F Bond Activation. J. Am. Chem. Soc. 2006, 128, 1806–1807. [Google Scholar] [CrossRef]
- Keen, A.L.; Doster, M.; Johnson, S.A. 1,4-Shifts in a Dinuclear Ni(I) Biarylyl Complex: A Mechanistic Study of C-H Bond Activation by Monovalent Nickel. J. Am. Chem. Soc. 2007, 129, 810–819. [Google Scholar] [CrossRef]
- Mukai, T.; Hirano, K.; Satoh, T.; Miura, M. Nickel-Catalyzed C-H Alkenylation and Alkylation of 1,3,4-Oxadiazoles with Alkynes and Styrenes. J. Org. Chem. 2009, 74, 6410–6413. [Google Scholar] [CrossRef] [PubMed]
- Tlili, A.; Schranck, J.; Pospech, J.; Neumann, H.; Beller, M. Ruthenium-Catalyzed Hydroaroylation of Styrenes in Water through Directed C-H Bond Activation. ChemCatChem 2014, 6, 1562–1566. [Google Scholar] [CrossRef]
- Thowfik, S.; Afsinab, M.A.; Anilkumar, G. Ruthenium-catalyzed hydroarylation reactions as the strategy towards the synthesis of alkylated arenes and substituted alkenes. RSC Adv. 2023, 13, 6246–6263. [Google Scholar] [CrossRef]
- Vechorkin, O.; Proust, V.; Hu, X. The Nickel/Copper-Catalyzed Direct Alkylation of Heterocyclic C-H Bonds. Angew. Chem. Int. Ed. 2010, 49, 3061–3064. [Google Scholar] [CrossRef]
- Ackermann, L.; Punji, B.; Song, W. User-Friendly [(Diglyme)NiBr2]-Catalyzed Direct Alkylations ofHeteroarenes with Unactivated Alkyl Halides through C-H Bond Cleavages. Adv. Synth. Catal. 2011, 353, 3325–3329. [Google Scholar] [CrossRef]
- Aihara, Y.; Chatani, N. Ruthenium-catalyzed direct arylation of C-H bonds in aromatic amides containing a bidentate directing group: Significant electronic effects on arylation. Chem. Sci. 2013, 4, 664–670. [Google Scholar] [CrossRef]
- Aihara, Y.; Chatani, N. Nickel-Catalyzed Direct Alkylation of C-H Bonds in Benzamides and Acrylamides with Functionalized Alkyl Halides via Bidentate-Chelation Assistance. J. Am. Chem. Soc. 2013, 135, 5308–5311. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L. Metal-catalyzed direct alkylations of (hetero)arenes via C-H bond cleavages with unactivated alkyl halides. Chem. Commun. 2010, 46, 4866–4877. [Google Scholar] [CrossRef] [PubMed]
- Chao Liu, C.; Dong Liu, D.; Wei Zhang, W.; Liangliang Zhou, L.; Lei, A. Nickel-Catalyzed Aromatic C-H Alkylation with Secondary or Tertiary Alkyl–Bromine Bonds for the Construction of Indolones. Org. Lett. 2013, 15, 6166–6169. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Lackner, S.; Ackermann, L. A General Strategy for the Nickel-Catalyzed C-H Alkylation of Anilines. Angew. Chem. Int. Ed. 2016, 55, 3153–3157. [Google Scholar] [CrossRef] [PubMed]
- Vijayarajan Devannah, V.; Watson, D.A. Nickel-Catalyzed C-Alkylation of Nitroalkanes with Unactivated Alkyl Iodides. J. Am. Chem. Soc. 2017, 139, 8110–8113. [Google Scholar] [CrossRef]
- Soni, V.; Jagtap, R.A.; Gonnade, R.G.; Punji, B. Unified Strategy for Nickel-Catalyzed C-2 Alkylation of Indoles through Chelation Assistance. ACS Catal. 2016, 6, 5666–5672. [Google Scholar] [CrossRef]
- Pandey, D.K.; Ankade, S.B.; Ali, A.; Vinod, C.B.; Punji, B. Nickel-catalyzed C-H alkylation of indoles with unactivated alkyl chlorides: Evidence of a Ni(I)/Ni(III) pathway. Chem. Sci. 2019, 10, 9493–9500. [Google Scholar] [CrossRef]
- Patel, U.N.; Pandey, D.K.; Rajesh, G.; Gonnade, R.G.; Punji, B. Synthesis of Quinoline-Based NNN-Pincer Nickel(II) Complexes: A Robust and Improved Catalyst System for C-H Bond Alkylation of Azoles with Alkyl Halides. Organometallics 2016, 35, 1785–1793. [Google Scholar] [CrossRef]
- Mandapati, P.; Braun, J.D.; Sidhu, B.K.; Wilson, G.; Herbert, D.E. Catalytic C-H Bond Alkylation of Azoles with Alkyl Halides Mediated by Nickel(II) Complexes of Phenanthridine-Based N^N–^N Pincer Ligands. Organometallics 2020, 39, 1989–1997. [Google Scholar] [CrossRef]
- Buendia, M.B.; Higginson, B.; Kegnæs, S.; Kramer, S.; Martin, R. Redox-Neutral Ni-Catalyzed sp3 C-H Alkylation of α-Olefins with Unactivated Alkyl Bromides. ACS Catal. 2022, 12, 3815–3820. [Google Scholar] [CrossRef]
- Sun, S.-Z.; Romano, C.; Martin, R. Site-Selective Catalytic Deaminative Alkylation of Unactivated Olefins. J. Am. Chem. Soc. 2019, 141, 16197–16201. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-B.; Zhao, X.; Zhang, D.; Sh, S.-L. Enantio- and Regioselective Ni-Catalyzed para-C-H Alkylation of Pyridines with Styrenes via Intermolecular Hydroarylation. J. Am. Chem. Soc. 2022, 144, 13643–13651. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Two representative transition-metal-catalyzed C-H bond alkylation strategies.

Scheme 2.
C-H bond alkylation of imidazolium salts.

Scheme 3.
Activation of a C-H bond in pentafluorobenzene.

Scheme 4.
Formation of branched 5-alkyl-2-phenyl-1,3,4-oxadiazoles.

Scheme 5.
Well-defined Ni(II)-complex in the 2-C-alkylation of N, O and S-containing heteroaromatics.
Scheme 5.
Well-defined Ni(II)-complex in the 2-C-alkylation of N, O and S-containing heteroaromatics.

Scheme 6.
Cooperation of nickel and copper in the 2-C-alkylation of azaheteroaromatics.

Scheme 7.
Mechanistic studies and possible pathway of nickel-catalyzed alkylation of fused azoles.

Scheme 8.
N,N-bidentate directing group in ortho-C-H bond alkylation of amides. a NaI (2 equiv.) added.
Scheme 8.
N,N-bidentate directing group in ortho-C-H bond alkylation of amides. a NaI (2 equiv.) added.

Scheme 9.
Simplified reaction pathway for the alkylation of the ortho-C-H bond through bidentate chelation assistance.
Scheme 9.
Simplified reaction pathway for the alkylation of the ortho-C-H bond through bidentate chelation assistance.

Scheme 10.
Intramolecular C-H alkylation with tertiary alkyl–Br bonds.

Scheme 11.
Intramolecular C-H alkylation with secondary alkyl–Br bonds.

Scheme 12.
Proposed mechanism for intramolecular C-H alkylation of N-aryl α-bromoamides.

Scheme 13.
Ortho-C-H alkylation of N-pyrimidylanilines.

Scheme 14.
Proposed mechanism for Ni-catalyzed C–H alkylation of 2-pyrimidinyl anilines with alkyl halides.
Scheme 14.
Proposed mechanism for Ni-catalyzed C–H alkylation of 2-pyrimidinyl anilines with alkyl halides.

Scheme 15.
Easy removal of the pyrimidyl directing group.

Scheme 16.
C-H bond alkylation of nitroalkanes with unactivated alkyl iodides.

Scheme 17.
Synthesis of adapromine by alkylation of nitroalkanes.

Scheme 18.
(Quinolinyl)amido-Ni(II) catalyst in the C-2 alkylation of indoles. a KI (2 equiv.) was added.
Scheme 18.
(Quinolinyl)amido-Ni(II) catalyst in the C-2 alkylation of indoles. a KI (2 equiv.) was added.

Scheme 19.
Synthesis of tryptamine alkaloid.

Scheme 20.
Directing group-assisted 2-C-H alkylation of indoles and pyrroles with primary chlorides.
Scheme 20.
Directing group-assisted 2-C-H alkylation of indoles and pyrroles with primary chlorides.

Scheme 21.
Directing group-assisted 2-C-H alkylation of indoles with secondary halides.

Scheme 22.
Proposed mechanism for Ni-catalyzed C-H alkylation of 2-pyridinyl indoles with alkyl halides.
Scheme 22.
Proposed mechanism for Ni-catalyzed C-H alkylation of 2-pyridinyl indoles with alkyl halides.

Scheme 23.
One-pot synthesis of bis(indolyl)alkane scaffolds.

Scheme 24.
Nickel(II) complexes of quinoline-based N,N,N-ligands in alkylation of benzothiazoles and benzoxazoles. a Using alkyl iodide instead of bromide without addition of NaI.
Scheme 24.
Nickel(II) complexes of quinoline-based N,N,N-ligands in alkylation of benzothiazoles and benzoxazoles. a Using alkyl iodide instead of bromide without addition of NaI.

Scheme 25.
Nickel(II) complexes of phenanthridine-based N,N,N-ligands in alkylation of benzoxazoles.
Scheme 25.
Nickel(II) complexes of phenanthridine-based N,N,N-ligands in alkylation of benzoxazoles.

Scheme 26.
Nickel catalysis in combination with iridium photocatalysis in coupling of α-olefins with primary alkyl bromides.
Scheme 26.
Nickel catalysis in combination with iridium photocatalysis in coupling of α-olefins with primary alkyl bromides.

Scheme 27.
Nickel catalysis in combination with metal-free photocatalysis in coupling of α-olefins with secondary alkyl bromides.
Scheme 27.
Nickel catalysis in combination with metal-free photocatalysis in coupling of α-olefins with secondary alkyl bromides.

Scheme 28.
Deaminative alkylation of unactivated α-olefins.

Scheme 29.
Deaminative alkylation of unactivated internal olefins.

Scheme 30.
(a) Deaminative one-pot alkylation without isolation of pyridinium salt. (b) Use of ethylene as substrate.
Scheme 30.
(a) Deaminative one-pot alkylation without isolation of pyridinium salt. (b) Use of ethylene as substrate.

Scheme 31.
Natural product derivatives obtained by deaminative alkylation in late-stage functionalization.
Scheme 31.
Natural product derivatives obtained by deaminative alkylation in late-stage functionalization.

Scheme 32.
Asymmetric addition of the pyridine ortho-C-H bond to styrenes.

Scheme 33.
Proposed mechanism for Ni-catalyzed C-H alkylation of pyridines with styrenes.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Požgan, F.; Grošelj, U.; Svete, J.; Štefane, B.; Al Mamari, H.H. Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds. Molecules 2024, 29, 1917. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091917
AMA Style
Požgan F, Grošelj U, Svete J, Štefane B, Al Mamari HH. Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds. Molecules. 2024; 29(9):1917. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091917
Chicago/Turabian StylePožgan, Franc, Uroš Grošelj, Jurij Svete, Bogdan Štefane, and Hamad H. Al Mamari. 2024. "Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds" Molecules 29, no. 9: 1917. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29091917