
Ligand Hydrogenation during Hydroformylation Catalysis Detected by In Situ High-Pressure Infra-Red Spectroscopic Analysis of a Rhodium/Phospholene-Phosphite Catalyst †
 ,
,  , ,
, ,
Abstract
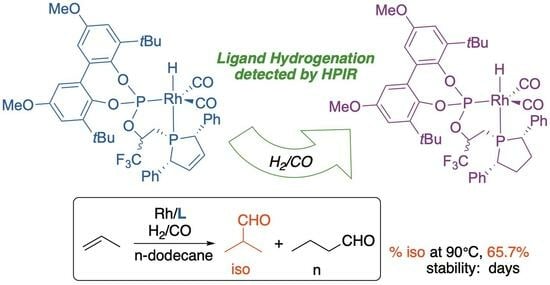
:
1. Introduction
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Safety Note
4.2. General Information
4.3. X-ray Crystallography
4.4. General Procedure for the Rhodium-Catalysed Hydroformylation of Propene
4.4.1. Synthesis of (Meso)-2,5-cis-diphenylphospholene borane Adduct 9: Borane-Protected (meso)-2,5-diphenyl-2,5-dihydro-1H-phosphole, 9
4.4.2. Borane-protected 3-((meso)-2,5-diphenyl-2,5-dihydro-1H-phosphol-1-yl)-1,1,1-trifluoropropan-2-ol, 10
4.4.3. 4,8-di-tert-butyl-6-((3-((meso)-2,5-diphenyl-2,5-dihydro-1H-phosphol-1-yl)-1,1,1-trifluoropropan-2-yl)oxy)-2,10-dimethoxydibenzo[d,f][1,3,2]dioxaphosphepine, 13a
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clark, T.P.; Landis, C.R.; Freed, S.L.; Klosin, J.; Abboud, K.A. Highly active, regioselective, and enantioselective hydroformylation catalysts ligated by Bis-3,3-diazaphospholanes. J. Am. Chem. Soc. 2005, 127, 5040–5042. [Google Scholar] [CrossRef]
- Zhang, W.; Chi, Y.; Zhang, X. Developing chiral ligands for asymmetric hydrogenation. Acc. Chem. Res. 2007, 40, 1278. [Google Scholar] [CrossRef]
- Xu, G.; Senayake, C.H.; Tang, W. P-chiral phosphorous ligands based on a 2.3-dihydrobenzo[d][1,3]oxaphosphole motif for asymmetric catalysis. Acc. Chem. Res. 2019, 52, 1101. [Google Scholar] [CrossRef]
- Ramazanova, K.; Chakrabortty, S.; Kallmeier, F.; Kretzschmar, N.; Tin, S.; Lönnecke, P.; De Vries, J.G.; Hey-Hawkins, E. The continued interest in chiral phosphacycles is evidenced by one of the papers in this special issue also being in this field. Molecules 2023, 28, 6210. [Google Scholar] [CrossRef]
- Axtell, A.T.; Klosin, J.; Abboud, K.A. Evaluation of asymmetric hydrogenation ligands in asymmetric hydroformylation reactions. Highly enantioselective ligands based on bis-phosphacycles. Organometallics 2006, 25, 5003–5009. [Google Scholar] [CrossRef]
- Carreira, M.; Charernsuk, M.; Eberhard, M.; Fey, N.; van Ginkel, R.; Hamilton, A.; Mul, W.P.; Orpen, A.G.; Phetmung, H.; Pringle, P.G. Anatomy of phobanes, Diasteroselective synthesis of three isomers of n-butylphobane and a comparison of their donor properties. J. Am. Chem. Soc. 2009, 131, 3078–3092. [Google Scholar] [CrossRef]
- Coles, N.T.; Abels, A.S.; Leitl, J.; Wolf, R.; Grützmacher, H.; Müller, C. Phosphinine-based ligands: Recent development in coordination chemistry and applications. Coord. Chem. Rev. 2021, 433, 213729. [Google Scholar] [CrossRef]
- Cobley, C.J.; Johnson, N.B.; Lennon, I.C.; McCague, R.; Ramsden, J.A.; Zanotti-Gerosa, A. Chap. 2 in Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions; Blaser, H.U., Schmidt, E., Eds.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Ager, D.J.; de Vries, A.H.M.; de Vries, J.G. Asymmetric homogeneous hydrogenations at scale. Chem. Soc. Rev. 2012, 41, 3340–3380. [Google Scholar] [CrossRef]
- Pilkington, C.J.; Zanotti-Gerosa, A. Expanding the family of phospholane-based ligands: 1,2-Bis(2,5-diphenylphospholano)ethane. Org. Lett. 2003, 5, 1273–1275. [Google Scholar] [CrossRef]
- Axtell, A.T.; Cobley, C.J.; Klosin, J.; Whiteker, G.T.; Zanotti-Gerosa, A.; Abboud, K.A. Highly Regio- and Enantioselective Asymmetric Hydroformylation of Olefins Mediated by 2,5-Disubstituted Phospholane Ligand. Angew. Chem. Int. Ed. 2004, 44, 5834. [Google Scholar] [CrossRef]
- Noonan, G.M.; Fuentes, J.A.; Cobley, C.J.; Clarke, M.L. An Asymmetric Hydroformylation Catalyst that Delivers Branched Aldehydes from Alkyl Alkenes. Angew. Chem. Int. Ed. 2012, 51, 2477–2480. [Google Scholar] [CrossRef]
- Pittaway, R.; Fuentes, J.A.; Clarke, M.L. Diastereoselective and branched-aldehyde-selective tandem hydroformylation–hemiaminal formation: Synthesis of functionalized piperidines and amino alcohols. Org. Lett. 2017, 19, 2845–2848. [Google Scholar] [CrossRef]
- Iu, L.; Fuentes, J.A.; Janka, M.E.; Fontenot, K.J.; Clarke, M.L. High iso aldehyde selectivity in the hydroformylation of short-chain alkenes. Angew. Chem. Int. Ed. 2019, 58, 2120–2124. [Google Scholar] [CrossRef]
- Herle, B.; Späth, G.; Schreyer, L.; Fürstner, A. Total Synthesis of Mycinolide IV and Path-Scouting for Aldgamycin N. Angew. Chem. Int. Ed. 2021, 60, 7893–7899. [Google Scholar] [CrossRef]
- Gilbert, S.H.; Fuentes, J.A.; Cordes, D.B.; Slawin, A.M.Z.; Clarke, M.L. Phospholane-phosphites for Rh catalyzed conjugate addition: Unsually reactive catalysts for challenging couplings. Eur. J. Org. Chem. 2020, 20, 3071–3076. [Google Scholar] [CrossRef]
- Ortiz, K.G.; Dotson, J.J.; Robinson, D.J.; Singman, M.S.; Karimov, R.R. Catalyst-controlled Enantioselective and Regiodivergent Addition of Aryl Boron Nucleophiles to N-Alkyl Nicotinate Salts. J. Am. Chem. Soc. 2023, 145, 11781–11788. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, J.A.; Janka, M.E.; Rogers, J.; Fontenot, K.J.; Bühl, M.; Slawin, A.M.Z.; Clarke, M.L. Effect of ligand backbone on the selectivity and stability of rhodium hydroformylation catalysts derived from phospholane-phosphites. Organometallics 2021, 40, 3966–3978. [Google Scholar] [CrossRef]
- Dingwall, P.; Fuentes, J.A.; Crawford, L.; Slawin, A.M.Z.; Bühl, M.; Clarke, M.L. Understanding a Hydroformylation catalyst that produces branched aldehydes from alkyl alkenes. J. Am. Chem. Soc. 2017, 139, 15921–15932. [Google Scholar] [CrossRef]
- Bagi, P.; Kovács, T.; Szilvási, T.; Pongrácz, P.; Kollár, L.; Drahos, L.; Fogassy, E.; Keglevich, G. Platinum,(II) complexes incorporating racemic and optically active 1-alkyl-3-phospholene P-ligands: Synthesis, stereostructure, NMR properties and catalytic activity. J. Organomet. Chem. 2014, 751, 306–313. [Google Scholar] [CrossRef]
- Leca, F.; Réau, R. 2-Pyridyl-2-phospholene: New P,N ligands for the palladium-catalyzed isoprene telomerisation. J. Catal. 2006, 238, 425–429. [Google Scholar] [CrossRef]
- Lin, J.; Coles, N.T.; Dettling, L.; Steiner, L.; Felix-Witte, J.; Paulus, N.M.; Müller, C. Phospholenes from Phosphabenzenes by Selective Ring Contraction. Chem. Eur. J. 2022, 28, e202203406. [Google Scholar] [CrossRef]
- Lim, K.M.-H.; Hayashi, T. Dynamic Kinetic Resolution in Rhodium-Catalyzed Asymmetric Arylation of Phospholene oxides. J. Am. Chem. Soc. 2017, 139, 8122–8125. [Google Scholar] [CrossRef]
- Hintermann, L.; Schmitz, M. Enantioselective Synthesis of Phospholene via Asymmetric Organocatalytic Alkene Isomerization. Adv. Synth Catal. 2008, 350, 1469–1473. [Google Scholar] [CrossRef]
- Hintermann, L.; Schmitz, M.; Maltsev, O.V.; Naumov, P. Organocatalytic stereoisomerization versus alkene isomerization: Catalytic asymmetric synthesis of 1-hydroxy-trans-2, 5-diphenylphospholane 1-oxide. Synthesis 2013, 45, 308–325. [Google Scholar] [CrossRef]
- Guillen, F.; Rivard, M.; Toffano, M.; Legros, J.-Y.; Daran, J.-C.; Fiaud, J.-C. Synthesis and first applications of a new family of chiral monophosphine ligand: 2,5-diphenylphosphospholanes. Tetrahedron. Tetrahedron 2002, 58, 5895–5904. [Google Scholar] [CrossRef]
- Quin, L.D.; Barket, T.P. Stereoisomerism in some derivatives of the 2-substituted 3-phospholene system. J. Am. Chem. Soc. 1970, 92, 4303–4308. [Google Scholar] [CrossRef]
- Esguerra, K.V.N.; Fall, Y.; Petitjean, L.; Lumb, J.-P. Controlling the catalytic aerobic oxidation of phenols. J. Am. Chem. Soc. 2014, 136, 7662–7668. [Google Scholar] [CrossRef]
- Byrne, J.J.; Chavant, P.Y.; Averbuch-Pouchot, M.-T.; Vallee, Y. 2,2’Biphenol. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 1154. [Google Scholar] [CrossRef]
- Elsler, B.; Schollmeyer, D.; Waldvogel, S.R. Synthesis of iodobiaryls and dibenzofurans by direct coupling at BDD anodes. Faraday Discuss. 2014, 172, 413–420. [Google Scholar] [CrossRef]
- Franke, R.; Selent, D.; Börner, A. Applied hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Börner, A.; Franke, R. Hydroformylation. Fundamentals, Processes, and Applications in Organic Synthesis; Börner, A., Franke, R., Eds.; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Puckette, T. Hydroformylation catalysis at Eastman chemicals. Top. Catal. 2012, 55, 421–425. [Google Scholar] [CrossRef]
- Guo, L.; Sun, L.; Huo, Y.X. Towards bioproduction of oxo chemicals from C1 feedstocks using isobutyraldehyde as an example. Biotechnol. Biofuels 2022, 15, 80. [Google Scholar] [CrossRef]
- Ibrahim, M.Y.S.; Bennett, J.A.; Mason, D.; Rodgers, J.; Abolhasani, M. Flexible homogeneous hydroformylation: On-demand tuning of aldehyde branching with a cyclic fluorophosphite ligand. J. Catal. 2022, 409, 105–117. [Google Scholar] [CrossRef]
- Wang, X.; Nurttila, S.; Czik, W.I.; Becker, R.; Rodgers, J.; Reek, J.N.H. Tuning the porphyrin building block in self assembled cages for branched selective hydroformylation of propene. Chem. Eur. J. 2017, 23, 14769–14777. [Google Scholar] [CrossRef]
- Sigrist, M.; Zhang, Y.; Antheame, C.; Dydio, P. Isoselective Hydroformylation by Iodide-Assisted Palladium Catalysis. Ange. Chem. Int. Ed. 2022, 61, e202116406. [Google Scholar] [CrossRef]
- How, R.C.; Dingwall, P.; Hembre, R.T.; Ponasik, J.A.; Tolleson, G.S.; Clarke, M.L. Composition of catalysts resting states of hydroformylation catalysts derived from bulky mono-phosphorous ligands, rhodium dicarbonyl acetylacetonate and syngas. Mol. Catal. 2017, 434, 116–122. [Google Scholar] [CrossRef]
- Chikkali, S.H.; van der Vlugt, J.I.; Reek, J.N.H. Hybrid diphosphorus ligands in rhodium catalysed asymmetric hydroformylation. Coord. Chem. Rev. 2014, 262, 1–15. [Google Scholar] [CrossRef]
- Castillo-Molina, D.A.; Casey, C.P.; Müller, I.; Nozaki, K.; Jäkel, C. New low temperature NMR studies establish the presence of Second equatorial-apical isomer of [(R,S)-BINAPHOS](CO)2RhH. C. Organometallics 2010, 29, 3362–3367. [Google Scholar] [CrossRef]
- Hommer, H.; Gordillo, B. Synthesis and study of the thermal epimerization of r-2-Ethoxy-cis-4-cis-5-Dimethyl-1,3,2-3-Dioxaphosholane using 31 P NMR. Phosphorous. Sulfur Silicon 2002, 177, 465–470. [Google Scholar] [CrossRef]
- Egan, W.; Tang, R.; Zon, G.; Mislow, K. Low barrier to pyramidal inversions in phospholes. Measure of aromaticity. J. Am. Chem. Soc. 1970, 92, 1442–1444. [Google Scholar] [CrossRef]
- Cremer, S.E.; Chorvat, R.J.; Chang, C.H.; Davis, D.W. Pyramidal inversion in substituted phosphetanes. Tetrahedron Lett. 1968, 55, 5799–5802. [Google Scholar] [CrossRef]
- Hoge, G. Stereoselective cyclization and pyramidal inversion strategies for P-chirogenic phospholane synthesis. J. Am. Chem. Soc. 2004, 126, 9920–9921. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, J.A.; Clarke, M.L. Ligand Hydrogenation during Hydroformylation Catalysis Detected by In-Situ High Pressure Infra-Red Spectroscopic Analysis of a Rhodium/Phospholene-Phosphite Catalyst: Dataset; University of St Andrews: St Andrews, UK, 2023. [Google Scholar]
- Allian, A.D.; Garland, M. Spectral resolution of fluxional organometallics: The observation and FTIR characterization of all-terminal [Rh4(CO)12]. Dalton. Trans. 2005, 2005, 1957. [Google Scholar] [CrossRef]
- Crystal Clear-SM Expert; Version 2.1; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2015.
- Crys Alis Pro; Version 1.171.42.96a; Rigaku Oxford Diffraction; Rigaku Corporation: Tokyo, Japan, 2023.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Crystal Structure; Version 4.3.0; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2018.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Li, C.; Xiong, K.; Yan, L.; Jiang, M.; Song, X.; Wang, T.; Chen, X.; Zhan, Z.; Ding, Y. Designing highly efficient Rh/CPOL-bp&PPh3 heterogeneous catalysts for hydroformylation of internal and terminal olefins. Catal. Sci. Technol. 2016, 6, 2143–2149. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13a | 14 | NEKXEJ | |
|---|---|---|---|
| Pd-P(1) | 2.2023 (5) | 2.183 (2) | |
| Pd-P(31) | 2.2426 (2) | 2.225 (2) | |
| Pd-Cl(2)trans phosphite | 2.3385 (5) | 2.342 (3) | |
| Pd-Cl(1)trans phosphine | 2.3271 (5) | 2.348 (2) | |
| P(1)-Pd-P(31) | 96.22 (2) | 86.13 (9) | |
| Cl(1)-Pd-Cl(2) | 92.50 (1) | 93.8 (1) | |
| O(2)-P1-O(13) | 106.47 (7) | 106.5 (4) | |
| C(2)-C(1)-C(12)-C(13) | 51.0 (2) | 65 (1) | |
| C(6)-C(1)-C(12)-C(17) | 45.0 (2) | 62 (1) | |
| Phenol-phenol twist | 53.05 (4) | 50.07 (6) | 66.8 (4) |
 | ||||
|---|---|---|---|---|
| Entry | Ligand [a] | T [°C] | TON (in 1 h) | Iso (%) |
| 1 [b] | 13a | 50 | 204 | 71.0 |
| 2 | 13a | 75 | 117 | 67.5 |
| 3 | 1 | 75 | 121 | 74.6 |
| 4 | 13a | 90 | 333 | 65.7 |
| 5 [c] | 1 | 90 | 397 | 70.9 |
| 6 | 13a | 105 | 751 | 64.5 |
| 7 | 1 | 105 | 782 | 67.2 |
| 8 | 13b | 75 | 78 | 67.0 |
| 9 | 13b | 90 | 184 | 64.8 |
| 10 | 13b | 105 | 502 | 62.3 |
| 11 | 13c | 75 | 181 | 65.3 |
| 12 | 13c | 90 | 462 | 63.3 |
| 13 | 13c | 105 | 993 | 61.0 |
| 11b | 13a | 14 | |
|---|---|---|---|
| formula | C48H50O2 | C41H45O5F3P2 | C44H48O5F3P2Cl11Pd |
| fw | 658.92 | 736.71 | 1272.11 |
| crystal description | colourless plate | colourless block | yellow prism |
| crystal size [mm3] | 0.1 × 0.1 × 0.02 | 0.12 × 0.11 × 0.04 | 0.1 × 0.09 × 0.05 |
| space group | P3221 | P21/c | P21/n |
| a [Å] | 9.9188 (3) | 18.5498 (3) | 14.94065 (19) |
| b [Å] | 10.34298 (16) | 19.3961 (2) | |
| c [Å] | 32.7080 (13) | 20.2110 (3) | 18.4869 (2) |
| β [°] | 103.8332 (16) | 95.1095 (11) | |
| vol [Å]3 | 2786.78 (16) | 3765.21 (11) | 5336.02 (11) |
| Z | 3 | 4 | 4 |
| ρ (calc) [g/cm3] | 1.178 | 1.300 | 1.583 |
| μ [mm−1] | 0.534 | 0.174 | 1.011 |
| F(000) | 1062.0 | 1552.0 | 2568.0 |
| reflections collected | 12302 | 74481 | 115446 |
| independent reflections (Rint) | 3712 (0.1250) | 9058 (0.0333) | 13030 (0.0352) |
| parameters, restraints | 235/1 | 468/27 | 753/166 |
| GoF on F2 | 1.225 | 1.028 | 1.031 |
| R1 [I > 2σ(I)]s | 0.0586 | 0.0386 | 0.0291 |
| wR2 (all data) | 0.2517 | 0.0997 | 0.0682 |
| largest diff. peak/hole [e/Å3] | 0.44/−0.30 | 0.30/−0.33 | 0.55/−0.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuentes, J.A.; Janka, M.E.; McKay, A.P.; Cordes, D.B.; Slawin, A.M.Z.; Lebl, T.; Clarke, M.L. Ligand Hydrogenation during Hydroformylation Catalysis Detected by In Situ High-Pressure Infra-Red Spectroscopic Analysis of a Rhodium/Phospholene-Phosphite Catalyst. Molecules 2024, 29, 845. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29040845
Fuentes JA, Janka ME, McKay AP, Cordes DB, Slawin AMZ, Lebl T, Clarke ML. Ligand Hydrogenation during Hydroformylation Catalysis Detected by In Situ High-Pressure Infra-Red Spectroscopic Analysis of a Rhodium/Phospholene-Phosphite Catalyst. Molecules. 2024; 29(4):845. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29040845
Chicago/Turabian StyleFuentes, José A., Mesfin E. Janka, Aidan P. McKay, David B. Cordes, Alexandra M. Z. Slawin, Tomas Lebl, and Matthew L. Clarke. 2024. "Ligand Hydrogenation during Hydroformylation Catalysis Detected by In Situ High-Pressure Infra-Red Spectroscopic Analysis of a Rhodium/Phospholene-Phosphite Catalyst" Molecules 29, no. 4: 845. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29040845